PROGERIA INFORMATION
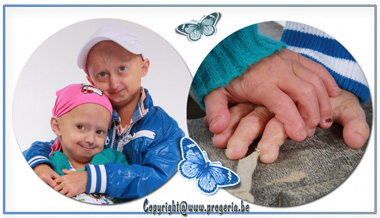
Race with time Too young to already be old A child is not just a thing, something bare. Deeply in that thing conceals something spectacular. There in lives a wonder. Something unbelievably powerful. They are much more then just splendid. We know what it is, and they make us stronger. Such a human being is not just man’s work. They are children like any other child, with just one exception. Michiel & Amber have Progeria. Their life may be short but this special young boy and girl, touches the hearts of everyone they meet. They have the same hopes and dreams that all children have.
1. Foreword :
On 13 June 1998 we became the proud parents of a little boy, Michiel. He suffers from the rare disease Progeria. On 5 February 2006 we became the proud and happy parents of a sweet little girl. Her name is Amber. When she was 6 weeks old, she was also diagnosed with Progeria. Michiel and Amber are the only sibling in the whole wide world who suffer from the classical form of Progeria “HGPS”. Because we have two children who suffer from this rare disease, Progeria, we will try to describe this deadly childhood disease and explain the aspects of it. Since we are daily directly involved, we have a whole lot of information that we want to share with you.
Our wish is to bring all the right information to the outside and to give greater publicity to Progeria. Our Amber is still small and young so therefore the information will more concern Michiel because we have more experience with him. Later on the information will be adapted to Amber as well. The environment of the parents and the child mostly doesn’t know how they must behave. The child sees after all totally different from the other children, eventually it can no longer play as other children (limited freedom of movement) and will die early. Also the support fails and does not give us the support that we, the parents, so need. Through this information we want to make everyone attentive on Progeria and the problems which a relatively unknown, rare illness bring with it.
2. Synonyms of “Progeria”:
- HGPS
- Hutchinson-Gilford Progeria Syndrome,
- Premature Aging Syndrome,
- Progeria of Childhood,
- Progeria Infantiles
3. Definition “Progeria” :
 Progeria is a rare, fatal, genetically determined disease of childhood characterized by dramatic, premature aging that occurs at about 8 to 10 times the normal rate of aging. Because of this accelerated aging, a child of ten years will have similar respiratory, cardiovascular, and arthritic conditions that a 70-year-old would have. Progeria is the most radical of the ageing illnesses. While there are different forms of Progeria*, the classic type is Hutchinson-Gilford Progeria Syndrome, which was named after the doctors who first described it in England; in 1886 by Dr. Jonathan Hutchinson and in 1904 by Dr. Hastings Gilford. Its name is derived from Greek and means “prematurely old”.
Progeria is a rare, fatal, genetically determined disease of childhood characterized by dramatic, premature aging that occurs at about 8 to 10 times the normal rate of aging. Because of this accelerated aging, a child of ten years will have similar respiratory, cardiovascular, and arthritic conditions that a 70-year-old would have. Progeria is the most radical of the ageing illnesses. While there are different forms of Progeria*, the classic type is Hutchinson-Gilford Progeria Syndrome, which was named after the doctors who first described it in England; in 1886 by Dr. Jonathan Hutchinson and in 1904 by Dr. Hastings Gilford. Its name is derived from Greek and means “prematurely old”.
Progeria occurs in approximately 1 in 4–8 million newborns, affecting both sexes equally and all races. One in 18-20 million living individuals has classic Progeria. According to the Progeria Research Foundation, as of June 30, 2023, there are worldwilde 198 identified children and young adults with Progeria living across 51 total countries. This figure includes 147 children with the classic Hutchinson-Gilford Progeria, who have a progerin producing mutation in the LMNA gene, and 51 children in the Progeroid Laminopathy category who have a mutation in the Lamin pathway but do not produce progerin. Amongst them 2 are living in Belgium and 1 in the Netherlands.
Children from all races and cultures around the world have been affected. In the past 15 years, children with Progeria have been reported all over the world, including in Algeria, Argentina, Australia, Austria, Belgium, Canada, China, Cuba, England, France, Germany, Israel, Italy, Mexico, Morocco, the Netherlands, Poland, Portugal, Puerto Rico, South Africa, South America, South Korea, Switzerland, Turkey, the US, Venezuela, Vietnam and Yugoslavia. Progeria signs include growth failure, loss of body fat and hair, aged-looking skin, stiffness of joints, hip dislocation, generalized atherosclerosis, cardiovascular (heart) disease and stroke. The children have a remarkably similar appearance, despite differint ethnic background. Without Zokinvy (lonafarnib) treatment, all children with Progeria die of the same heart disease that affects millions of normally aging adults (arteriosclerosis), but at an average age of just 14.5 years.
Most children with Progeria don’t live beyond their early teenage years, though one or two have lived to their early 20s. It is always assumed that Progeriachildren never come into puberty. However, we disagree because Michiel is experiencing his puberty. He has the same dreams and desires as boys of his age. Progeriachildren develop no sex hormones. Girls with Progeria can have their periods. Because there are so few children known with this rare illness, not many physicians know the syndrome from their medical practice.
Therefore:
- the illness sometimes becomes recognized very late,
- Progeria children are misdiagnosed by physicians
- A child despite all good intentions, has been subjected to rather aggravating treatments which have not helped.
In Belgium (Flanders) there are 2 children/young adults having the condition ‘Progeria’ : Michiel (23 years) & Amber (15 years). Unfortunately our dear friend Toon died at the age of 12 years on November 9, 2014, and our beloved Mats died at the age of 17 on December 18, 2019.

Passport Michiel Vandeweert : 
- Date of birth: 13/06/1998
- Horoscope : Twins
- Weight: 20 kg.
- Length: 129,5 cm.
- Hobbies: Playing soccer, computer games, horse around with someone, playing D.J.
- Colors of his eyes: blue
- Character: Social, Charming, Curious, Enthusiastic, Friendly, Funny, Happy, Loving, Social, Spontaneous, Sweet, Stubborn, Assertive, Naughty, Honest, Playful
Passport Amber: 
- Date of birth: 05 / 02 / 2006
- Horoscope: Aquarius
- Weight: 17 kg.
- Lenght: 123 cm.
- Hobbies: shopping, making TikTofmovies, meeting up with friends
- Colors of her eyes: Brown
- Character: fond of laughing, roguish, playful
Passport Toontje* (deceased on November 9, 2014): 
- Date of Birth: 03 / 03 / 2002
- Horoscope : Fish
- Weight: 9,650 kg.
- Length: 90 cm..
- Hobbies: Since I can walk, discovering the world, playing with mummy, daddy & my little brothers Stafke and Kamiel
- Colors of his eyes: Blue – grey
- Character: Enthusiastic, Social, Spontaneous, fond of laughing
Passport Mats* (deceased on December 18, 2019) : 
- Date of Birth: 09 / 09 / 2002
- Horoscope: Virgin
- Weight: 23kg
- Length: 130,5 cm.
- Hobbies: get into mischief, horse around, playing outside, jumping on the trampoline, …
- Colors of his eyes: Brown
- Character: always ready for a joke, kind, strong personality, social, brave, …
4. Cause :
In 2003, a French medical team led by Dr. Nicolas Levy (Marseille) and researchers of the NIH in the US have discovered the cause of HGPS. The driving force within this American team is Dr. Leslie Gordon. HGPS is caused by a mutation in the gene called LMNA (pronounced, lamin – a). The LMNA gene produces the Lamin A protein, which is the structural scaffolding that holds the nucleus of a cell together. Researchers now believe that the defective Lamin A protein makes the nucleus unstable. That cellular instability appears to lead to the process of premature aging in Progeria.
It is caused by a point-mutation in Chromosome 1. This causes a defect in the production of proteins Lamin A and C. In the DNA of these children the place of a genetic failure: this fault acts ‘ spontaneously ‘ at conception. By a deviation to LMNA-gen (the Lamine A) the body of Progeria patients can not produce Lamin-a well. Because of this the inside of the nucleus of a cell becomes abnormally composed. HGPS can be diagnosed by looking at the specific genetic change, or mutation, in the Progeria gene that leads to HGPS. After an initial clinical evaluation (looking at the child’s appearance and medical records), a sample of the child’s blood will be tested for the Progeria gene. For the first time ever, there is a definitive, scientific way to diagnose the children. This will lead to more accurate and earlier diagnoses so that the children can receive proper care.
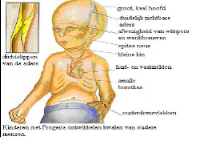
5. Symptoms / characterize:
Although they are born looking healthy, children with Progeria begin to display many characteristics of accelerated aging at around 18-24 months of age. The course of classical Progeria is for all children almost the same. The average birth weight is about three kilos and they are approximately 50 cm. long. The child does however not grow sufficient. At birth, Michiel had three spots in his face and a complete dry belly. The three spots has disappeared in the next days. His dry belly has remained. Amber had no spots at birth nor a dry skin. A first symptom is the striking visible vein concerning the nose bridge, sometimes also on the forehead. 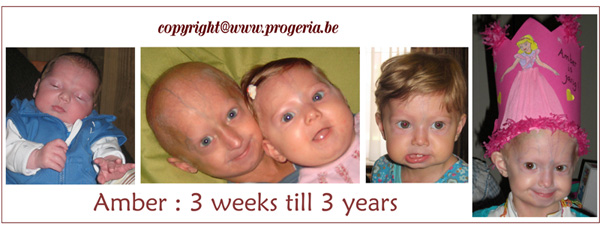 In the first year, the following arises:
In the first year, the following arises: 
- stagnation in growth, dwarfed growth; serious growth delay;
- beginning hair loss;
- changes of the breast and belly skin: Leathery skin, dry and sometimes red in color, formation of fat edema (scleroderma);
- strong growth of the skull size
- decreased agility
The further development of the child lapses till at the end of the first year normal and discreetly.
Image from the second year: 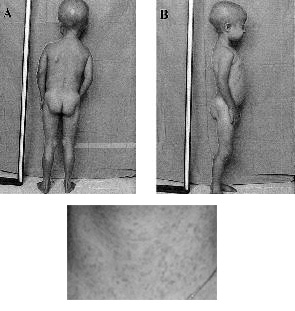
- Lively children that however increase scarcely in weight and length.
- the children appear fragile.
- the small face with the large, rather round eyes is striking.
- a relatively small jaw in which the teeth often have too little space.
- Thin or mostly no head hair
- Thin, tight and translucent skin.
- The skin has little elasticity, and therefore extra vulnerable.
- clear visible blood vessels, particularly on the head..
- circles under the eyes.
- hyperpigmentations (lentigo senilis)
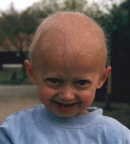

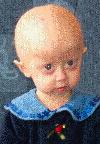
Image after the two first years:
- striking short and broad nails and shortened last bones in one’s finger (acro-osteolysis);
- subcutaneous body fat disappears;
- extraordinary position of the legs : slightly stiff with light bent knees. With help of X-rays is to be seen that the position of the upper leg points outward (coxa valga).
- the tendons no longer will develop. They will move stiff with light bent knees
- stunted growth of collarbones; also using X-rays to determine
- bothered by an increasing stiffness of the joints (arthritis). Usually begins in the fingers and knees, wooden movements. Also they can suffer from rheumatic disorders.
- Changes of the blood vessels and of all other organs, arise through the advancing arterioscleroses resulting in bleeding disorders and connective tissue scleroses in the body.
- Decreased senses
- diabetes
- hip dislocation
- Thin, high-pitched voice
- Protruding ears; lack of ear lobes
 Finally culminating with the following:
Finally culminating with the following:
- Heart and vascular diseases
- Arteriosclerosis
- Heart Attack(s)
- cerebral infarction & strokes: brain hemorrhages
Michiel and Amber had all the symptoms as described from the first and second life year. Collage Michiel
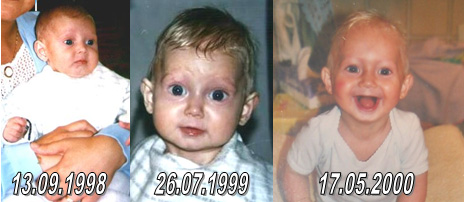
Collage Amber
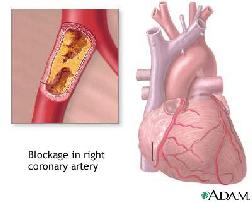
 Reduced mobility and vascular disorderproblems cost much energy to the child. Progeriachildren can act only short periodes a day as a normal child because they are quickly tired. The most important health problems which follow concern heart- and blood vessels. Progeriachildren can already get a cerebral infarction at the age of 4 or 5 years wiith consequences on their mobility and speech capacity. Fortunately that does not happen frequently. The heartproblems,however, are frequent. They generally start between the 8th and 12e year. They gradually becomes worse. Sometimes heartcomplaints appear also acute. As with any person suffering from heart disease, the common events for Progeria children are high blood pressure, strokes, angina (chest pain due to poor blood flow to the heart itself), enlarged heart, and heart failure, all conditions associated with aging.
Reduced mobility and vascular disorderproblems cost much energy to the child. Progeriachildren can act only short periodes a day as a normal child because they are quickly tired. The most important health problems which follow concern heart- and blood vessels. Progeriachildren can already get a cerebral infarction at the age of 4 or 5 years wiith consequences on their mobility and speech capacity. Fortunately that does not happen frequently. The heartproblems,however, are frequent. They generally start between the 8th and 12e year. They gradually becomes worse. Sometimes heartcomplaints appear also acute. As with any person suffering from heart disease, the common events for Progeria children are high blood pressure, strokes, angina (chest pain due to poor blood flow to the heart itself), enlarged heart, and heart failure, all conditions associated with aging.
- Length – weight:
- The average length of Progeriakids of 12 years can vary between 96 cm. and 128 cm. They seldom become larger than 115 cm. The weight of a twelve year old one lies between 9.3 kilogram. up to 20.7 kilogram.
- Growth:
- Progeriachildren grow too slow. Often the child eats worse. Both the increase in weight and in length remains behind but the weight increases the less. There will be no increase in the growthprocess in the prepuberty nor puberty.
- Hair:
- The little hair of the head is thin and fluffy. They get downy hair. Also the hair of eyebrows and eye members disappears almost entirely.
- Skin:
- The skin is thin, tightly and translucent. There are, particularly on the head, clear visible blood vessels. Beside it develop lentigo senilis (hyperpigmentations).
- Loss of subcutaneous fat and clear visible blood vessels :
- Loss of subcutaneous fat can already appear from the age of six months, but becomes generally just visible around three to four-year age. This is first to see at the limbs, after that at the chest and the skull and finally in the face. Up to that time the children often have chubby bloom cheeks. The subcutaneous layer of fat of stomach and hips disappears last. The fat in the belly itself however does not disappear therefore the often swollen belly of Progeriachildren. By the disappearance of subcutaneous fat and the getting thinner of the skin, the veins become more clearly visible. Because the face fat also disappears, the eyes seem slightly round.
- Teeth:
- Beside it many progeriachildren have irregular teeth. The teeth and molars do not have sufficient space in the jaw to grow tidy beside each other. The teeth and molars are coming through late. With most of the children the first teeth come through between 8 and 24 months. Dental caries comes often through. Cleaning theeth is hard in such a small mouth but also the children who brush their teeth well, in generally have quickly problems with dental caries.
- Heart- and blood vessels:
- Initially the children have no problems with the heart- or the blood vessels. Also the blood pressures is normal in the first years. Of four years, mostly between six and eight years, heart murmer can be heard.From the sixth up to eighth year the children can become short of breath, with high blood pressure. X-rays sometimes shows that the heart is slightly enlarged. Angina pectoris frequently appears as well as arteriosclerosis. The vascular problems can reach everywhere in the body: tightness of the chest and breathing problems appear and also a cerebral infarction is caused by silted up veins. Some children recover entirely, others keep problems after having a cerebral infarction.
- Skeleton and joints :
- One of the first symptoms of Progeria is the very late or not closing of the cranial sutures. This applies especially the fore fontanelle, which can remain even open till puberty. The crown of the skull is largely depending on the growth of the brains and because the brains at Progeria continue to grow, the crown of the skull stays normal to big. This, together with the boneloss of the facebones and small jaw, is the cause that the skull comparatively speaking is very large regard to the face. Because the jaw gets smaller, the teeth and molars can become standing higgledy-piggledy In the beginning the muscles are well visible by the disappearance of subcutaneous fat. The legs look muscular. Later also the muscle mass decreases. The shoulder blades are well visible, they stick out. By artrose the mobility of the joints decreases increasingly further, also those in ankles, wrists, shoulders and hip. As a result, the joints become painful. A shortening of (one of) the neck muscles can appear and many children get a slightly convex back.
6.Inheritance :
The chance that a Progeria child gets a little brother or sister with the same illness is quite small. There are on these two exceptions: the American identical twins (boys) Chris and Chad (1978) and the Malaysian identical twins (girls) Lim Sook Khuen and Lim Sook Wen. Meanwhile the 4 children passed away. Now there are also the first non-twin sibling : Michiel and Amber (Below on photograph)  Children with other types of “progeroid” syndromes which are not HGPS may have diseases that are passed down in families. However, Progeria is an autosomal dominant ageing disease which is caused by an sporadic mutation in the LMNA-gene. This means that, for a family with one child with HGPS, non-twin siblings have the same chance of having HGPS as any other child in any other family. When a couple gets a child with a deviant gene, then it is possible that this mutation has arisen at several moments. This can be happened during the formation of that one sex cell from which the child has arisen, but also earlier (in the parent) or later (in the child). With Progeria the mutation generally arises after the conception, in the child.
Children with other types of “progeroid” syndromes which are not HGPS may have diseases that are passed down in families. However, Progeria is an autosomal dominant ageing disease which is caused by an sporadic mutation in the LMNA-gene. This means that, for a family with one child with HGPS, non-twin siblings have the same chance of having HGPS as any other child in any other family. When a couple gets a child with a deviant gene, then it is possible that this mutation has arisen at several moments. This can be happened during the formation of that one sex cell from which the child has arisen, but also earlier (in the parent) or later (in the child). With Progeria the mutation generally arises after the conception, in the child.
The classical type of Progeria, HGPS, the child does not inherate (in principle) of the parents, because she arises nearly always by a spontaneous mutation during one of the first cell divisions. In very rare instances HGPS can be passed down within a family. When the mutation has arisen earlier, a number of cells contains the mutation of the concerning parent. Since Amber’s Birth and by research (father and mother) one knows that the classical type HGPS can be determined by heredity (by an inherited mutation). In our extremely rare case the mother of the child carries the LMNA-mutation in her sex cells, without her having problems of them. This parent (mother) has a mixture of cells with and without the mutation; this also is called a “mosaic”. Depending on when the mutation has appeared, the number of cells with the mutation at the parent will vary. The mosaic can because of this be present in several tissues, or only in the sex cells. When one of the parents carries the mutation in the sex cells, the chance gets larger that the child will have Progeria. Germline mosaicism is possible. In a patient with a classical p.G608G mutation, the phenotypically normal mother was found to have a somatic mosaicism, 10% of her buccal cells harboring the same mutation [Wuyts et al., 2005].
She must have had a germ line mosaicism too. Prenatal testing is available, however, because of the low risk of recurrence, prenatal testing would only be performed because of the (limited) possibility of germ line mosaicism (two or more genetic or cytogenetic cell lines confined to the precursor (germline) cells of the egg or sperm; formerly called gonadal mosaicism) in one of the parents. Progeria patients hardly ever get the chance to reproduce. They often die prematurely, and besides that the disrupted hormonal development prevents the Progeria patient from becoming fertile. If they would have the possibility to reproduce, they would have a large possibility that their children would also have Progeria. In India there is a family with 5 Progeriachildren, namely the children Kahn. However, they do not suffer from the classical form of Progeria but to a variant of it. You see also clearly the differences between all children with HGPS (our children) and the children Kahn:
- children with classical Progeria (such as our children): Thin, high-pitched voice, voice will never breaking, bald from 2 years, smal head, pointed nose, little small teeth, heart problems are cause of death number 1, our children have a remarkably similar appearance .
- children Kahn: one boy his voice was breaking, a deep voice, they all had hair (even the children of 15 and 19 years), round faces, large broad teeth and they had no problems with the heart.
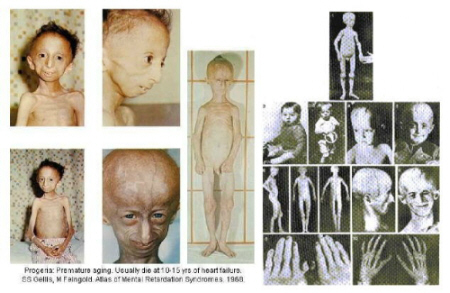
At classical progeria ‘HGPS” heartfailure is he most important cause of death. Nine year old American boy, Ory Barnett got a heart attact at the age of 5. Nevertheless, the faith of the Children Kahn is as worse as that of our children. 7. X-Rays: (left : normal person / right : Progeriachild)

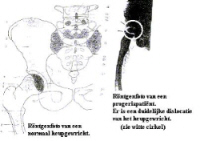

X-Rays : FALLER A., Het menselijke lichaam (bouw en functie), Oosthoek, Utrecht, 1975
8. Physical and mental development:
The physical consequences of Progeria are not yet preventable. Progeria is incurable. Medical science tries only to treat it’s symptoms. Through vasoconstriction, there is a strong increased risk of problems at young age with saturated blood in the brains or a heart attack. To reduce these risks a low daily dose aspirin is prescribed. Despite the many agreements, the illness develops differently with every child. By strengthen the complete constitution of the child with for example feeding supplements, life quality will improve. The intellectual development of children with Progeria developes normally. They have no intellectual deprivation.
At school they are bright and inquisitive children. Children with Progeria develop their skills just like their same age companions. However, most children with progeria have a problem developing their fine motor skills. Michiel has also problems with fine motor skills: for example he doesn’t like to write or paint because he gets cramps in his fingers. Since he knows the alphabet, he uses an especially for him designed computer. It is however no game computer. With this he can follow in class (does not get behind) and he can follow the tempo. School tasks or house work are made on this computer by him.  Despite everything, almost all children with Progeria live their life with heart and soul : they begin each new day full of joy and they enjoy all the beautiful things that life has to offer. They have a sunny view on life, are full of life, and thanks to their open, kind and high-spirited nature, they soon are fully accepted by their classmates. By their unusual appearance, children with Progeria are stared at; A situation, with which they and their parents must learn to deal with. They react ad-rem and humoristic to the sometimes tactless remarks. They find fast friends and girlfriends at school.
Despite everything, almost all children with Progeria live their life with heart and soul : they begin each new day full of joy and they enjoy all the beautiful things that life has to offer. They have a sunny view on life, are full of life, and thanks to their open, kind and high-spirited nature, they soon are fully accepted by their classmates. By their unusual appearance, children with Progeria are stared at; A situation, with which they and their parents must learn to deal with. They react ad-rem and humoristic to the sometimes tactless remarks. They find fast friends and girlfriends at school.
9. Emotional process :
The diagnosis of Progeria is a huge shock for family members . Sometimes years go by before the diagnosis is accepted and before living with Progeria can be experienced as positive. It is a difficult situation to deal with the perspective, the physical consequences of the illness and the sometimes blunt reactions from the surroundings. BUT THE MOST IMPORTANT THING OF COURSE IS THAT THE CHILD IS FULLY ACCEPTED AND FEELS THAT HE IS LOVED. Unconditional love is indispensable for these children. The child must have the opportunity to act and behave as a normal child. For the child, it can be hard to accept the illness, its consequences and the pain.
Most (older) children pass away fully knowing what is going on. They realize it quite well. These children and their parents sometimes have many questions without answers. After the diagnosis the parents have a difficult problem: how can you live with this illness and accept it? Despite the sorrow, can you focus on the development possibilities of your child? That is the first and most difficult problem for the family after the diagnosis Progeria. A painful and emotional time follows. We are forced to revise our performances and expectations of life. As a father or mother you wrestle with questions about problems that in the course of time will come. And concerning all questions that the child will ask you, you must answer truthfully and in a manner that fits with the age of the child. By that, you teach the child to accept itself.
Also it is very important that the child feels independence and has a feeling of self-respect. Michiel wants to do everything independently; he doesn’t want help so we had to do some necessary adaptations in our home, for example : put the doorhandles lower, make all the lights with a cord on the end, put an additional stair handrail to his length, a step on the bath and toilet, placed a fitted washbasin at his height and so on.
Adaptions in house 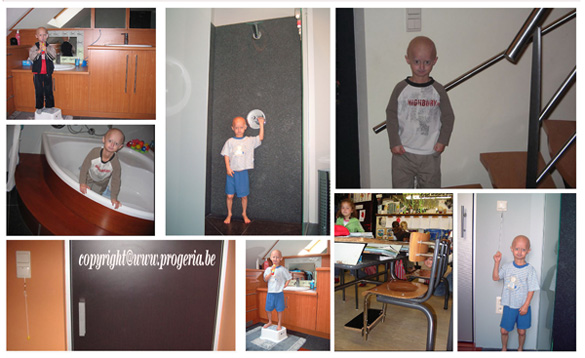 Expectations change. With Progeria expectations concerning “Later, I will be large” , are no longer possible. The child can do less and less. There are no expectations in the long term and parents no longer want to think in the long term. They want to enjoy each and every day. The parents live day by day and take every day as it comes. No planning long term. Step by step. By the illness Progeria, the care always remains. During a time the care will be less but it will increase when the children will get more problems. The greatest aid for a child with Progeria is of course the help of the parents who fully support their child.
Expectations change. With Progeria expectations concerning “Later, I will be large” , are no longer possible. The child can do less and less. There are no expectations in the long term and parents no longer want to think in the long term. They want to enjoy each and every day. The parents live day by day and take every day as it comes. No planning long term. Step by step. By the illness Progeria, the care always remains. During a time the care will be less but it will increase when the children will get more problems. The greatest aid for a child with Progeria is of course the help of the parents who fully support their child.
But where can the parents turn to for help ? When you as a parent need emotional support, who will take care of you ? Is there somebody you can turn to ? Are there people willing to help you or where you get help with your questions ? It is therefore a long, emotional and uncertain process for the parents. They need the support of others (partner, family, friends, companions, neighbours, …). They undergo a very difficult stage in their life, but they have no other choice than to deal with it. Most of the parents accept it after a time of course, by the aid of others. They must finally give this a place in their heart otherwise it is unbearable. And of course they still have their good and bad moments. If there is a day that one parent has difficulties, they must be able to say this to the other parent so that he/she can give a hug or a pep talk; or they may isolate and cry (with or without each other). The parents don’t have to be strong the entire time; there must be space to give your sorrow the upper hand. This was also the case for Godelieve and Wim. They could turn to family, friends etc. But they had the need to talk with somebody who fully understands everything about Progeria, giving up a child to this rare disease.
So they found Marjet Stamsnijder, the chairwoman of the Progeria Family Circle. She introduced them to other families with a child that suffers from Progeria It is necessary to know that you can rely on others because you receive no psychological help after the diagnosis. You receive the diagnosis by the professor and that is it. You are on your own at these moments. The psychological help that you really need, is lacking and very limited. Since you have an enormous need for support, you seek it somewhere else : your partner, your sister, your brother, your best friend or family. You seek someone with which you feel secure and with whom you can express your mixed feelings and sorrow. But the period before the diagnosis has been already terrible for the parents.
There was the uncertainty and when the final diagnosis was made, our world was broken down completely and all hope fell away.The fact is, that once the diagnosis is said aloud and you know that your child will die early, you have to contend with different feelings : mainly grief, denial, helplessness, confusion, anger, despair, depression. Some even have feelings of guilt. By giving greater publicity to Progeria, the parents no longer need to walk fearful on the street. Passers-by no longer look at them as if their son/daughter has a narrow illness. Everywhere in the world they make Progeria widely known. Firstly passers-by thought for instance that Michiel had a contagious disease and they walked around him in large arcs. Nowadays people know better. Their children may play with these children and the parents are encouragingly addressed. Michiel is a beautiful example of this. He has become through his acquaintance inhabitant of Limburg of the year 2003. One would not have chosen him if they had found him scary; no they admire him.

Finally, these children have the same needs as those of an ordinary child; they also play with pleasure, they want to do certain things like other children, for example : Michiel loves to play soccer so he joined a soccer team but he can not play the whole match (quickly tired, gets cold very fast, …). There are certain restrictions in the development of the child. Michiel plays with his friends or schoolmates but when he is tired, he takes a break and participates later on. They all understand that. Michiel can swim entirely independently. That is a complete step ahead and we are of course very proud of this performance.
Because he has no subcutaneous fat, he cools down quickly in water (his lips and hands colors blue). Because of this we have bought him a wet suit so he can stay longer in the water. He tries everything he likes at his own speed. Because they look differently than other children, they are frequently gaped at or they get negative and hurtful remarks. People would rather avoid these children than laugh at them and this is what the parents try to prevent by bringing Progeria to the forefront and giving people the opportunity to become acquainted with this disease. This was also the case for Michiel. People looked at him oddly and didn’t let their children near him because they were afraid he was contagious. Everywhere we went, people stared at him (even with open mouths), whispering about him and pointing at him.
This hurt Godelieve terribly. Acceptance in society was still somewhat difficult. Therefore, Godelieve and Wim decided to do something about this and they used the media to gain acceptance for Michiel and his fellow-sufferers and they managed garner more respect and understanding. Once his story was in the media, people were sympathetic & understanding and even offered their aid and assistance.
Many families find that once they do television shows or documentaries, people will recognize the child and be more compassionate towards the child. All these children have a pleasant, cheerful and social character and they are easygoing in their contact with others. Since the articles in the newspapers and the programs on television our life has become more beautiful. People no longer stare at him but cheer him and that let extra sunshine into our hearts.
That is what we needed : understanding, sympathy and accepting Michiel like he is. Contact with other families with Progeria kids : Many times, the parents of children with Progeria feel alone in their battle because not many families deal with Progeria. In Belgium we were the first family with such a child. The annual meetings with other families dealing with Progeria has proven to be extremely important. They are a source of information as well as a source of support.
The contact with other children and their parents always gives much needed emotional support, parents find answers to their questions and get aid and assistance where possible. It allows parents to share their struggles with other adults who are experiencing the same type of situation. The hard part of the annual reunion is the realization that there probably will be some children who will not be able to return the following year.

Mutual contact is a great source for a better quality of life. Children with Progeria build up a lot of self-confidence and zest for living when meeting with each other. I honestly must admit that the very first confrontation, especially with the older children was very emotional. I cried continually. The parents each told their story. Each more terrible than the other. Some children were teased at school, thrown in the garbage can because they look so “different”,and some already had heart complaints.
And the beautiful story of Burçu who told us that she had the opportunity to meet her Turkish idol because of her disease. If she hadn’t had Progeria, she may never have had the chance to meet him. We also saw the ageing which was very noticeable and we suddenly became aware of Michiels situation. We were fully aware of what was in store for Michiel. Looking at them was suddenly seeing Michiel in the future. It was tremendously difficult.
You ask yourself a thousand and one questions, like : will he also have such a burden of subcutaneous pain? Will he just as Menekçe come to a point that he can no longer sit or stand by himself? Will he have an early burden on his heart? Have diabetes? A toothache? All scenarios overtake you in your sleep, you see the faces of the children in your dreams and you see the sorrow of the parents. You hear a child of 16 say that she has had more pain in her life than she has been happy; hearing that hurts a lot. But it is reality. During that first meeting with all the children, I had it very tough emotionally, but in spite of it all I did not want to miss it.
When we have a reunion, it feels like a homecoming , the children are not a curiosity and they see that they are not alone in being bald and small and having visible veins on their heads. The following weeks were emotionally very difficult for me until I could see it all in the right perspective. In spite of the first emotional confrontation, we looked forward to the next meeting. In the meantime we all have been together several times. The very first time I saw all the children, I was fearful but the second time the fear was already gone. When we all are together during a reunion, we are all one big happy family. There are strong bonds between all the families in spite of different nationalities, backgrounds and especially the language. Together we laugh, cry, and talk but most of all we enjoy without care.
All these children have a special place in our heart. They seem to have a magnetic personality that draws people to them. They all have the special gift to cheer somebody up , they are very spontaneous, social, fond of laughing and full of life. We no longer look at their appearance (external features) because their inner self is so special. They are exceptional children.  With our experience, we hope we can be a support for Liesbeth and Gerry and for the parents of Mats. There is contact between the three Belgian families, and that is very important.
With our experience, we hope we can be a support for Liesbeth and Gerry and for the parents of Mats. There is contact between the three Belgian families, and that is very important.
We all need this. Through our children, there is a special bond for life. Also we have contact with Bjorn and his family and we regularly see Hayley and her family and that is nice. They can call on us for aid, assistance, guidance just as we can ask for aid, assistance and guidance of the other families which have a child suffering from Progeria. With each other, we can exchange experiences but mostly we can bond friendships.
10. Treatments:
The following measures, derived from individual experiences of families, have turned out to be very helpful:
- Early doses of homeopathic or biochemical medicines. This method delays the effects of the arteriosclerosis and reduces pain and strained mental conditions.
- Early contact with other affected families and qualified therapists, so the children and their families receive emotional support, important information and help.
- Gentle physiotherapy and a lot of physical movement. This fights against premature stiffening of the joints. The muscles are strengthened and the blood circulation is promoted. Many children attend specialized physical therapy in order to keep their muscles and joints as supple and strong as possible. Many of the children find swimming/hydrotherapy beneficial as it helps relax the muscles and keeps them flexible.
- Good dental care of the first teeth and regular visits with the dentist. Treatments must be measured carefully here. There is often only a partial growth of second teeth, and the teeth are crowded. In the small jaw, there is mostly little space for the teeth, yet one must be careful with radical straightening of the teeth.
- Effective medical bath essences and body lotions help to alleviate the itch and to diminish the dryness of the thin skin.
- Timely acquisition of necessary aids. For example, the child may want a wig if the child goes to school.
- For poor eaters a food supplements is given when the calorie need of the child is larger then what is eaten. For instance the pediatrician or dietitian can prescribe as an addition to the usual daily meals Pediasure or Ensure.
- Purchase of necessary aids. Some children want to go independently to school so they need an adapted bicycle : for children with joint problems or a decreased heart position.
- Adaptations in the house: for instance an accessible door handle or a low sink promote the autonomy of the child, and promote a feeling of self-esteem.
11.Treatment :
A) Boston:
May 7, 2007 – December 2009: First-Ever Progeria Clinical Drug Trial Marks Historic Moment in Progeria Research History! Lonafarnib After only 7 years in existence, we became scientifically ready to begin this trial with a drug that shows great promise to effectively treat children with Progeria, and in the process we may help millions of older people who suffer from heart disease and other, aging-related conditions. Researchers have now identified a potential drug treatment for children with Progeria, called FTIs. For the first time, we have in front of us a possible treatment for children with Progeria. Exciting times! The Progeria clinical drug trial began on May 7th, 2007 with two children arriving in Boston, MA for their first of seven visits over a 2-year period. At this first visit, they were given extensive tests and their first doses of the drug. An average of two families have been flying to Boston each week since then, and in October 2007, the trial became fully enrolled.
The trial is expected to end in October 2009, with results published in 2010. Twenty-eight (28) children from sixteen countries are participating, ages 3 to 15 years. Children return to Children’s Hospital Boston every four months, for testing and to receive new drug supply, and stay in Boston for 4-8 days each visit. While at home, their doctors keep a close watch over the children and submit monthly health reports to the Boston research team. For the duration of the trial, 1- 2 children per week will travel to Boston to participate. Children originate from the following countries : Argentina, Belgium, Canada, Denmark, England, India, Israel, Italy, Japan, Mexico, Pakistan, Poland, Portugal, Romania, USA, Venezuela. Since the gene discovery, the support of researchers, clinicians, families of children with Progeria have brought us to another crossroads in the search for a treatment. Researchers have identified a potential drug treatment for children with Progeria, called farnesyltransferase inhibitors (FTIs), and have conducted studies in the lab that support a human trial with the drug.
The protein that we believe is responsible for Progeria is called progerin. In order to block normal cell function and cause Progeria, a molecule called a “farnesyl group” must be attached to the progerin protein. FTIs act by blocking (inhibiting) the attachment of the farnesyl group onto progerin. So if the FTI drug can block this farnesyl group attachment in children with Progeria, then progerin may be “paralyzed” and Progeria improved. Progeria cells become normalized when FTIs are applied. Capell et al., PNAS, 2005
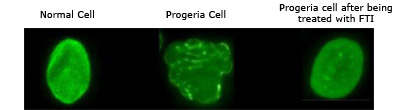
B) Marseille (France):
Juli 2008 : New hope for children with Progeria : Combined treatment with statins (Vasten®) and aminobisphosphonates (Zometa®) extends longevity in a mouse model of human premature aging Several human progerias, including Hutchinson-Gilford progeria syndrome (HGPS), are caused by the accumulation at the nuclear envelope of farnesylated forms of truncated prelamin A, a protein that is also altered during normal aging. Previous studies in cells from individuals with HGPS have shown that farnesyltransferase inhibitors (FTIs) improve nuclear abnormalities associated with prelamin A accumulation, suggesting that these compounds could represent a therapeutic approach for this devastating progeroid syndrome3.
We show herein that both prelamin A and its truncated form progerin/LA 50 undergo alternative prenylation by geranylgeranyltransferase in the setting of farnesyltransferase inhibition, which could explain the low efficiency of FTIs in ameliorating the phenotypes of progeroid mouse models. We also show that a combination of statins and aminobisphosphonates efficiently inhibits both farnesylation and geranylgeranylation of progerin and prelamin A and markedly improves the aging-like phenotypes of mice deficient in the metalloproteinase Zmpste24, including growth retardation, loss of weight, lipodystrophy, hair loss and bone defects.
Likewise, the longevity of these mice is substantially extended. These findings open a new therapeutic approach for human progeroid syndromes associated with nuclear-envelope abnormalities. Fifteen European children have started their treatment in Marseille. Toontje and Mats, the two other Belgian progeriakids from Belgium are also participating in this treatment. See the publication in Nature Medicine: http://www.nature.com/nm/journal/vaop/ncurrent/abs/nm1786.html
C) Boston : @ Progeria Research Foundation
August 2009 :
Announcing the Progeria Triple Drug Trial
 The Progeria Research Foundation and Children’s Hospital Boston are once again partnering to conduct a second clinical trial for children with Progeria. This exciting and much larger trial will include up to 45 children from 19 different countries! Researchers have identified two additional drugs that, when used in combination with the current FTI drug being tested, may provide an even more effective treatment for children with Progeria than FTI’s alone. Progeria is caused by an abnormal protein named progerin.
The Progeria Research Foundation and Children’s Hospital Boston are once again partnering to conduct a second clinical trial for children with Progeria. This exciting and much larger trial will include up to 45 children from 19 different countries! Researchers have identified two additional drugs that, when used in combination with the current FTI drug being tested, may provide an even more effective treatment for children with Progeria than FTI’s alone. Progeria is caused by an abnormal protein named progerin.
The Progeria research team at Children’s Hospital Boston will add two drugs, called pravastatin and zoledronate, to the current treatment with FTI. All three drugs will target different points along the pathway leading to production of the disease-causing progerin. In exciting laboratory studies presented by Dr. Carlos Lopez-Otin of Spain at the 2007 Progeria Research Foundation Scientific Workshop, the two new drugs improved disease in Progeria cells and extended lifespan in mouse models of Progeria. Goal: If the three drugs administered in this trial can effectively block this farnesyl group attachment, then progerin may be “paralyzed” and Progeria may be improved.
We hope that the drugs will work as partners, to complement each other so that the progerin protein is affected more by combining the three drugs than using any one drug alone
Who Will Enroll In The Triple Drug Trial? The Feasibility Trial: The team has already conducted a mini-trial for 5 children with Progeria. The short, one month “feasibility” trial, asked whether the three-drug combination would be well-tolerated, prior to embarking on a larger international trial. Side effects were acceptable, and the team has moved ahead to the larger efficacy trial.
The Efficacy Trial: We anticipate up to 45 children will enroll in this trial, from 19 different countries, speaking 11 different languages. This includes children participating in the FTI-only trial, the 5 in the feasibility trial, and other children that were either too young to participate in the first trial or children that we’ve discovered over the past 2 years. Children currently enrolled in the FTI-only trial will have the opportunity to enroll in the triple trial when they arrive for their last visit for the current trial. This allows the children to continue taking FTI without any missed doses. Trial Medications at a Glance Pravastatin (marketed as Pravachol or Selektine) is a member of the drug class of statins. It is usually used for lowering cholesterol and preventing cardiovascular disease. Zoledronic acid is a bisphosphonate, usually used as a bone drug for improving osteoporosis, and to prevent skeletal fractures in people suffering from some forms of cancer. Lonafarnib is an FTI (Farnesyltransferase inhibitor), a drug that can reverse an abnormality in Progeria cells in the laboratory, and has improved disease in Progeria mice. All 3 drugs block the production of the farnesyl molecule that is needed for progerin to create disease in Progeria. Patients will travel to Boston for testing and examinations lasting 4-7 days, every 6 months for a period of 2 years.
For the FTI-only trial, Boston visits occur every four months. In March, 2009, five children, ages 2-3, participated in a one-month feasibility study to determine if the side effects of the three drugs taken together were tolerable. The results were positive, paving the way for the full, two-year, Triple Drug Trial to enroll up to 45 children with Progeria. Hats off to these amazing families!
D) June 2011: PRF-funded study Identifies Rapamycin as Possible Treatment for Progeria : @Progeria Research Foundation :
Researchers at the National Institutes of Health and Massachusetts General Hospital in Boston, MA published a new study today in Science, Translational Medicine that may lead to a new drug treatment for children with Progeria.* 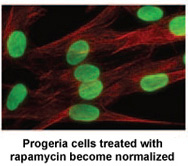 Rapamycin is an FDA approved drug that has previously been shown to extend the lives of non-progeria mouse models. This new study demonstrates that rapamycin decreases the amount of the disease-causing protein progerin by 50%, improves the abnormal nuclear shape, and extends the lifespan of progeria cells. This study provides the first evidence that rapamycin may be able to decrease progerin’s damaging effects in children with progeria. There is tremendous media coverage on this! Click below for links to media stories:
Rapamycin is an FDA approved drug that has previously been shown to extend the lives of non-progeria mouse models. This new study demonstrates that rapamycin decreases the amount of the disease-causing protein progerin by 50%, improves the abnormal nuclear shape, and extends the lifespan of progeria cells. This study provides the first evidence that rapamycin may be able to decrease progerin’s damaging effects in children with progeria. There is tremendous media coverage on this! Click below for links to media stories:
The Progeria Research Foundation was delighted to provide cells for this project from the PRF Cell & Tissue Bank, and help fund the research through our grants program. This exciting new study demonstrates the remarkable pace of progeria research, while providing further insight into the aging process that affects us all. *”Rapamycin Reverses Cellular Phenotypes and Enhances Mutant Protein Clearance in Hutchinson-Gilford Progeria Cells” Kan Cao, John J. Graziotto, Cecilia D. Blair, Joseph R. Mazzulli, Michael R. Erdos, Dimitri Krainc, Francis S. Collins 29 June 2011 Vol 3 Issue 89 Epub ahead of print.
Trials history at-a-glance:
Trial 4 began in April 2016 and is ongoing. It is a 2-drug, phase 1 trial to determine the maximum tolerated dose (MTD) of the drug everolimus, which is being taken along with the treatment lonafarnib. Once the MTD is determined, PRF will fund and co-coordinate the phase 2 portion, which will test effectiveness of the 2-drug combination.
New Drug, New Hope for Children with Progeria: Phase 1, 2-Drug Trial Begins April 2016
PRF is thrilled to announce that we are now funding and co-coordinating a new clinical trial, which will assess a two-drug combination of lonafarnib plus everolimus. Everolimus is a form of the drug rapamycin, but everolimus can be more easily given to the children with Progeria because it requires fewer blood draws to measure drug levels. While lonafarnib may block progerin from developing, rapamycin appears to allow cells to more rapidly clear out the toxic progerin. Thus with rapamycin targeting a different pathway than lonafarnib, the combination may prove to be a “one-two punch” to Progeria – hopefully a better treatment than lonafarnib on its own.
Rapamycin is an FDA-approved drug that has previously been shown to extend the lives of non-Progeria mouse models. A study* by researchers at the NIH in Bethesda, MD and Massachusetts General Hospital in Boston demonstrates that rapamycin decreases the amount of the disease-causing protein progerin by 50%, improves the abnormal nuclear shape, and extends the lifespan of Progeria cells in the laboratory.
Rapamycin is known for its anti-aging properties in mice. These findings are part of a growing list of studies that help to validate the theory that finding the cure for Progeria may also benefit the entire aging population.
* K. Cao, J. J. Graziotto, C. D. Blair, J. R. Mazzulli, M. R. Erdos, D. Krainc, F. S. Collins, “Rapamycin Reverses
Cellular Phenotypes and Enhances Mutant Protein Clearance in Hutchinson-Gilford Progeria Syndrome Cells.” Sci. Transl. Med. 3, 89ra58 (2011).
The Boston trial team will begin with Phase 1, where the study team will determine the safest maximum dose of everolimus for children with Progeria. The team begins by giving three children a very low dose of the medication, and carefully observes for side effects. If toxicity is minimal, another three children are enrolled at a higher dose of medicine. This pattern recurs until the safest maximum dose of everolimus is determined and the next study phase can begin. During Phase 2, the study will determine if the effects of the two-drug combination on disease are better than lonafarnib alone. Together, this Phase 1-2 treatment trial may enroll up to 80 children, and take an estimated 3.5-4 years to complete, at a cost of $2.5 million dollars.
The Progeria Research Foundation provided cells for this project from the PRF Cell & Tissue Bank and helped fund the research through our grants program – more proof that PRF’s research-related programs are essential to advancements toward the cure.
This new trial is a collaborative effort that will build upon the knowledge gained from the previous Progeria trials. The children will be seen by virtually the same team of physicians from Boston Children’s Hospital, Dana-Farber Cancer Institute and Brigham and Women’s Hospital, all of whom now have world-renowned expertise in Progeria as well as the drugs involved.
August 2017:
Michiel and Amber have been to Amerika in August 2017 and both have started with this new cure, which will last 2 years.
12. Classic and Non-classic Progeria :
Children with non-classic forms also do not resemble each other as closely as HGPS patients do, and they may also experience other complaints.
-
The lag in growth is less severe: children with non-classic Progeria grow 1.30 m to 1.45 m, while Progeriakids hardly grow taller than 1.15 m.
- The hair on the head is kept longer and does not disappear completely when they are older.
- Subcutaneous fat is not lost as quickly, and the cheeks and the skin under the chin retain their subcutaneous fat longer. This only disappears at an adult age.
- Bone loss is more visible than it is with Progeria, and occurs in many bones such as in the crown of the skull, jaw, collarbone, finger segments, and ribs. It also occurs in the facial bones (viscerocranium), but only later, at mature age. With much bone loss there is an elevated risk of bone fractures. These children tend to have many fractures as a result, often from the age of 3 or 4 years.
- A certain level of blood relation in the marriage of the parents appears to be more frequent (in the study by Dr. Hennekam, 4 of the 13 families).
- Chance of reaching adult age is higher than for Progeria.

Related diseases : The cause of Progeria lies on chromosome 1, in the gene that is involved in constructing the nuclear lamina. This is also the cause for a number of related diseases.
Dilated Cardiomyopathy: This is a condition weakening the heart muscles. In dilated cardiomyopathy, it is a disease of the heart muscle itself that leads to a diminishing of the pumping function. The course of the disease is slowly progressive that in general leads to worsening of the condition, and ultimately leads to heart failure.
Partial lipodystrophy: In this disorder patients have a deficiency of subcutaneous fat. The characteristics of the disorder are more easily seen with women than with men.
MAD, Mandibulo Acral Dysplasia : Mandibulo Aacral Dysplasia is a rare autosomal recessive syndrome.
Werner Syndrome: Werner syndrome is also an ageing-syndrome. In this disorder, which is hereditary, symptoms and complaints that are normally associated with old age arise earlier than normal. The disorder can first present itself at the onset of puberty, but also earlier or later. The average life-expectancy is 20-30 years.
Website: Progeria Family Circle http://www.progeria.nu/index2.html Progeria Family Circle : European Organization for Children with Progeria. Because the sickness Progeria is so rare, often children in Europe don’t know about each other’s existence. For this reason the European Progeria Family Circle was set up in 1997. They want to help Progeria Children and their families, support them and offer them information. This Progeria Family Circle decided also to organize Progeria meetings in Europe. To this day proving the importance of meeting each other and being involved in common activities. The children always have much pleasure and they also develop a stronger feeling of self-respect. Once they are together, they talk, laugh, cry, play etc.
For personal contact with the Progeria Family Circle, you can turn to: Marjet Stamsnijder PROGERIA FAMILY CIRCLE Foundation for children with Progeria Nude 6 3911 VK Rhenen The Netherlands Phonenumber : +31 (0)317 615391 E-mail address: progeriafamilycircle@hetnet.nl
The Sunshine Foundation : www.sunshinefoundation.org The Sunshine Foundation is a Make-A-Wish-type Organization which runs the Annual Progeria Reunion. An event that lets Progeria Children from around the world spend time with others like themselves. In 1981, the Sunshine Foundation had been in existence for five years and had handled approximately 500 special dreams of seriously ill, chronically ill, and physically challenged children.
It was brought to Sunshine’s attention in a newspaper article that a young boy in South Africa, named Fransie, had a dream to visit Pinocchio. The inspiration for this dream came from this boy seeing himself in a mirror. Because of his wooden appearance, he felt in some way related to Pinocchio.
Having read the article, Sunshine embarked on making yet another dream come true, unaware that this particular endeavor would bring about the knowledge of a very rare illness to the world. Fransie had his dream come true in December 1981. The Sunshine Foundation flew Fransie and his family to Disneyland where he saw Pinocchio. During his visit to the United States, three other children, also suffering from Progeria, met with him.
Progeria Research Foundation : www.progeriaresearch.org The Progeria Research Foundation raises money and researches Progeria’s cause as well as possible treatments and cures.
Hutchinson-Gilford Progeria Syndrome Resource Center : www.hgps.net One of the first sites with information about Progeria.
This is a source of photographs, media on TV, FAQ, links and even a forum. Europrogeria : www.europrogeria.org The first European symposium, aimed at presenting a clinical and molecular update of HGPS, was scheduled to coincide with this. During this symposium (from 25th September – 29th September 2003 in Magdeburg, Germany), a number of European experts in the field of laminopathies as well as clinicians with experience in the diagnosis and treatment of progeria and/or progeria-like syndromes and in relevant research presented their experiences. A decision was also made by the participants to work closely together in the field of progeria research. As a consequence, EuroProgeria, the European network for the scientific investigation of Hutchinson-Gilford Progeria, was established.
13. Decision:
We gladly want to decide with next words: Michiel is humorous, spontaneous, self-assured, stubborn, enthusiastic, social, but a GREAT child. He has developed into a self-confident and cheerful child, despite his rare illness. He goes to school and despite his self-willed personality, he is always surrounded by boy- and girlfriends who fully except him as he is. How can it be anything else? He is a child full of energy and humour and with his joy of living, he makes quite an impression on people, even strangers. He is the pride and joy of us all. Amber is a sweet cute little baby girl who has given us already so much joy. She is playfull, enchantig and an arch child. Michiel and Amber are the sun and warmth in our lifes. They teach us the real meaning of life : joy, love, togetherness and a tolerance for those who are different. ! ! MICHIEL AND AMBER CONTRIBUTES ENORMOUSLY TO OUR QUALITY OF LIFE !
————-
Sources / documentation : Wim + Godelieve Vandeweert / Marjet Stamsnijder / Progeria Family Circle / Progeria Research Foundation (Hennekam RC (2006). “Hutchinson-Gilford Progeria Syndrome: review of the phenotype”) : Click here Progeria Research Foundation : Clinical trial: Click here First-Ever progeria clinical drug trial reaches 1 year: Click here Announcing the Progeria Triple Drug Trial: Click here Hutchinson-Gilford Progeria Syndrome : GENEReviews : Click here