INFORMATIE PROGERIA
1. Voorwoord :
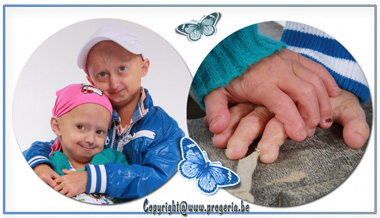
Op 13 juni 1998 werden wij de fiere en gelukkige ouders van Michiel. Hij lijdt aan de zeldzame ziekte Progeria. Op 5 februari 2006 werden wij opnieuw de trotse en gelukkige ouders van een lief klein meisje. Wij hebben haar Amber genoemd. Toen ze zes weken oud was, werd ook bij haar de diagnose van “Progeria” gesteld. Michiel & Amber zijn de enige broer en zus in de hele wereld die lijden aan de klassieke Progeria. Aangezien wij dus twee kinderen hebben, die lijden aan deze zeldzame ziekte “Progeria”, gaan wij deze dodelijke kinderziekte beschrijven en de aspecten ervan uitleggen. Daar wij er dagelijks rechtstreeks bij betrokken zijn, hebben wij inmiddels een hele hoop informatie die wij met jullie willen delen. Onze betrachting is immers om de juiste informatie naar buiten te brengen en om Progeria meer bekendheid te geven. Onze Amber is nog klein en daardoor zal de informatie nu nog meer gaan over Michiel aangezien wij met hem meer ervaring hebben. Later zal de informatie dan ook aangepast worden aan Amber. De omgeving van de ouders en het kind weet vaak niet hoe zij zich moeten gedragen. Het kind ziet er immers totaal anders uit dan andere kinderen, kan uiteindelijk niet meer zo spelen als andere kinderen en zal ook nog op vrij jonge leeftijd komen te overlijden. Ook de hulpverlening schiet vaak tekort en blijkt niet altijd de steun te geven die wij, de ouders, zo nodig hebben.
2. Synoniemen voor “Progeria” :
- HGPS
- Hutchinson-Gilford Progeria Syndrome
- Premature Aging Syndrome
- Progeria of Childhood
- Progeria Infantiles
3. Definitie “Progeria” :
 Letterlijk betekent Progeria “versneld ouder worden”. De klassieke vorm van progeria staat bekend als het Hutchinson-Gilford Progeria Syndroom (HGPS), genoemd naar de artsen Jonathan Hutchinson (in 1886) en Dr. Gilford Hastings (1904). Deze artsen hebben Progeria voor de eerste keer beschreven. Sinds 1886, het jaar waarin Progeria voor het eerst werd beschreven, zijn in totaal ongeveer 250 kinderen met Progeria beschreven. Kinderen met Progeria ontwikkelen voortijdig verouderingssymptomen die normaal gesproken alleen voorkomen bij oude mensen. Progeria is een genetisch bepaalde aandoening, en de meest ingrijpende onder de verouderingziekten. Kinderen met Progeria worden elk kalenderjaar ongeveer 8 tot 10 levensjaren ouder. De gemiddelde levensverwachting is 14,5 jaar. Slechts enkele kinderen zijn rond hun 20ste levensjaar overleden. Kinderen en jongvolwassenen met Progeria ontwikkelen geen geslachtshormonen. Meisjes met Progeria kunnen ongesteld worden. Er werd altijd aangenomen dat deze kids niet in hun puberteit komen. Echter kunnen wij dat hier tegenspreken want Michiel beleeft wel zijn puberteit : hij zit met dezelfde dromen en verlangens als jongens van zijn leeftijd.
Letterlijk betekent Progeria “versneld ouder worden”. De klassieke vorm van progeria staat bekend als het Hutchinson-Gilford Progeria Syndroom (HGPS), genoemd naar de artsen Jonathan Hutchinson (in 1886) en Dr. Gilford Hastings (1904). Deze artsen hebben Progeria voor de eerste keer beschreven. Sinds 1886, het jaar waarin Progeria voor het eerst werd beschreven, zijn in totaal ongeveer 250 kinderen met Progeria beschreven. Kinderen met Progeria ontwikkelen voortijdig verouderingssymptomen die normaal gesproken alleen voorkomen bij oude mensen. Progeria is een genetisch bepaalde aandoening, en de meest ingrijpende onder de verouderingziekten. Kinderen met Progeria worden elk kalenderjaar ongeveer 8 tot 10 levensjaren ouder. De gemiddelde levensverwachting is 14,5 jaar. Slechts enkele kinderen zijn rond hun 20ste levensjaar overleden. Kinderen en jongvolwassenen met Progeria ontwikkelen geen geslachtshormonen. Meisjes met Progeria kunnen ongesteld worden. Er werd altijd aangenomen dat deze kids niet in hun puberteit komen. Echter kunnen wij dat hier tegenspreken want Michiel beleeft wel zijn puberteit : hij zit met dezelfde dromen en verlangens als jongens van zijn leeftijd.
Progeria komt voor bij ongeveer 1 op de 4–8 miljoen pasgeborenen en treft beide geslachten in gelijke mate en alle rassen. Een op de 18-20 miljoen levende individuen heeft klassieke Progeria. Volgens de Progeria Research Foundation zijn er vanaf 30 juni 2023 wereldwijd 198 geïdentificeerde kinderen en jongvolwassenen met Progeria bekend in 51 landen. Hiervan zijn er 147 met de klassieke vorm van HGPS en 51 anderen met een variante. Van deze kinderen en jongvolwassenen met Progeria wonen er 2 in België en 1 in Nederland. In de afgelopen 15 jaar werden er kinderen met Progeria gerapporteerd over de hele wereld waaronder Algerije, Argentinië, Australië, België, Canada, China, Cuba, Engeland, Frankrijk, Duitsland, Israël, Italië, Mexico, Marokko, Nederland, Oostenrijk, Polen, Portugal, Puerto Rico, Zuid Afrika, Zuid-Amerika, Zuid-Korea, Zwitserland, Turkije, Verenigde Staten, Venezuela, Vietnam en Joegoslavië. Er zijn dan ook niet veel artsen die het syndroom uit hun medische praktijk kennen. Progeria is heel zeldzaam, maar een ziekte die overal ter wereld voor komt, ongeacht geslacht, ras of huidskleur.
Daardoor wordt :
- de ziekte soms zeer laat erkend,
- er vaak een verkeerde diagnose gesteld,
- een kind ondanks alle goede bedoelingen, nogal eens aan belastende behandelingen onderworpen waar het niet mee geholpen is.
In België (Vlaanderen) zijn enkel Michiel (23) en Amber (15) die Progeria hebben, deze zeldzame ziekte. Ons dierbare vriendje, Toon is helaas op 12-jarige leeftijd overleden op 9 november 2014. Onze dierbare Mats is op 17-jarige leeftijd overleden op 18.12.2019.

Paspoort Michiel: 
- Geboortedatum: 13/06/1998
- Horoscoop: Tweeling
- Gewicht: 20 kg.
- Lengte: 1 meter 29,5 cm.
- Hobby’s: voetbal, ravotten, computerspelletjes spelen, D.J.
- Kleur ogen: Blauw
- Mijn karakter: Enthousiast, Sociaal, Spontaan, Eigenzinnig, Assertief, Ondeugend, Eerlijk, Charmant, Speels, Koppig, Goedlachs, sterke persoonlijkheid
Paspoort Amber: 
- Geboortedatum: 05 / 02 / 2006
- Horoscoop: Waterman
- Gewicht: 17 kg.
- Lengte: 1 meter 23 cm.
- Hobby’s: Shoppen, TikTokvideos opnemen, afspreken met vriendinnen
- Kleur ogen: Bruin
- Mijn karakter: Goedlachs, guitig, speels, levenslustig, ondeugend, sterke persoonlijkheid
Paspoort Toontje* (overleden op 09.11.2014): 
- Geboortedatum: 03 / 03 / 2002
- Horoscoop: Vis
- Gewicht: 9,650 kg.
- Lengte: 90 cm.
- Hobby’s: Met mama en papa spelen, stoeien met Stafke en Kamiel
- Kleur ogen: Blauw-grijs
- Karakter: Enthousiast, Sociaal, Spontaan, goedlachs
Paspoort Mats* (overleden op 18.12.2019): 
- Geboortedatum: 09 / 09 / 2002
- Horoscoop: Maagd
- Gewicht: 23kg
- Lengte: 1 meter 30,5 cm
- Hobby’s: Kattekwaad uithalen, ravotten, buiten spelen, trampoline springen
- Kleur ogen: Bruin
- Karakter: altijd te vinden voor een grapje, lief, stoer, sterke persoonlijkheid, moedig, sociaal
4. Oorzaak :
De oorzaak van Progeria is in 2003 aangetoond door een Frans medisch team onder leiding van Dr. Nicolas Lévy in Marseille, en door onderzoekers van het NIH in de VS. De drijvende kracht binnen dit Amerikaanse team is Dr. Leslie Gordon. Het genetisch defect treedt waarschijnlijk spontaan op bij één van de eerste celdelingen na de conceptie. Het gaat om een eenmalige, spontaan voorkomende verandering van een erfelijke factor; een mutatie. Het genetisch defect in het DNA is gelokaliseerd op chrosomoom 1, en wordt de G608G mutatie genoemd. Deze kleine afwijking op het LMNA-gen veroorzaakt vroegtijdige verouderingsverschijnselen bij kinderen met Progeria, doordat het eiwit aan de binnenzijde van de celkern niet volledig kan worden opgebouwd. Het lichaam van kinderen en jongvolwassenen met Progeria kan 15 aminozuren, die normaliter worden ‘opgeruimd’ in het opbouwproces, niet afbreken, omdat de code daarvoor op het gen ontbreekt. Dit onvolledige eiwit in de celkern heeft men progerin genoemd. Het veroorzaakt ‘blebbing’ van de cel en het verkort de celleeftijd sterk. Het progerin tast de gladde spieren en het epitheel van de bloedvaten aan. Hierdoor ontstaan hart- en vaatziekten, die ook de belangrijkste doodsoorzaak vormen. De diagnose van HGPS kan met zekerheid worden vastgesteld door een genetische test. Maar het is ook heel goed om te weten dat er zo hard wordt gewerkt en men steeds weer een stapje dichter bij een mogelijke therapie komt.
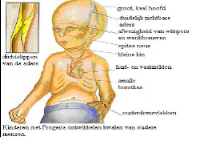
5. Symptomen / kenmerken :
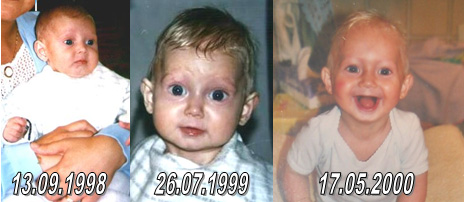
Het verloop van klassieke Progeria is bij alle kinderen nagenoeg gelijk. Het gemiddelde geboortegewicht is ongeveer drie kilo maar het kind komt niet voldoende bij en groeit niet zoals het hoort. Michiel had bij de geboorte drie vlekken in zijn gezicht en een hele droge huid op de buik. De drie vlekken zijn in de eerstvolgende dagen verdwenen. De huid op zijn buik is gebleven. Amber had bij haar geboorte geen vlekken op haar gezicht noch een droge huid. Een eerste symptoom is de opvallend zichtbare ader over de neusbrug, soms ook op het voorhoofd. 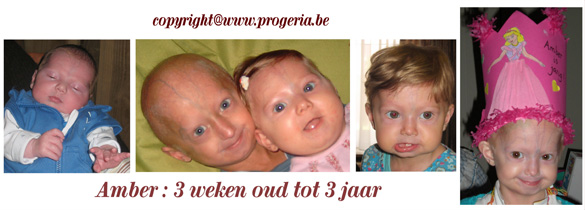 In het eerste levensjaar ontstaat het volgende beeld:
In het eerste levensjaar ontstaat het volgende beeld: 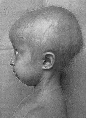
- stagnatie in groei, dwerggroei, ernstige groeivertraging;
- beginnende haaruitval;
- veranderingen van de borst- en buikhuid : droog en soms roodgekleurd, vorming van vetoedemen (sclerodermie);
- soms lijkt er even sprake te zijn van een verhoudingsgewijs sterke groei van de schedelomvang;
- motorisch wat verminderde beweeglijkheid
De verdere ontwikkeling van het kind verloopt tot aan het einde van het eerste levensjaar normaal en onopvallend.
- levendige kinderen die echter nauwelijks in gewicht en lengte toenemen;
- de kinderen maken een fragiele indruk;
- opvallend is het kleine gezichtje met de grote, enigszins bolle ogen;
- een relatief kleine kaak waarin de tanden vaak te weinig ruimte hebben;
- weinig, dun, meestal geen hoofdhaar;
- dunne, strakke en doorschijnende huid;
- De huid is weinig elastisch,en daardoor extra kwetsbaar;
- duidelijk zichtbare bloedvaten, met name op het hoofd;
- kringen onder de ogen;
- ouderdomsvlekjes.
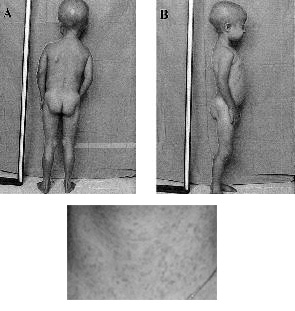
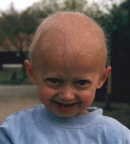

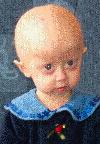
Beeld na de twee eerste levensjaren:
- opvallend korte (brede) nageltjes en verkorte laatste vingerkootjes (acro-osteolysis);
- lichaamsvet verdwijnt;
- afwijkende stand van de benen; enigszins stijf met licht gebogen knieën. Met behulp van röntgenfoto’s is te zien dat de stand van het bovenbeen naar buiten gedraaid is (coxa valga).
- hun pezen gaan zich ook niet meer ontwikkelen, waardoor ze zich stijf gaan bewegen met licht gebogen knieën
- niet volledig volgroeide sleutelbeenderen; ook met behulp van de röntgenfoto’s vast te stellen;
- een toenemende stijfheid van de gewrichten (artritis, artrose), die meestal begint in de vingergewrichten en de knieën : bewegingen worden houterig. Ook kunnen er reumatische klachten ontstaan.
- veranderingen aan de bloedvaten en aan alle andere organen, ontstaan door de voortschrijdende arterosclerose, met als gevolg doorbloedingsstoornissen en bindweefselverhardingen in het lichaam.
- verminderde zintuigen
- diabetes
- heupontwrichting
- hoog stemmetje
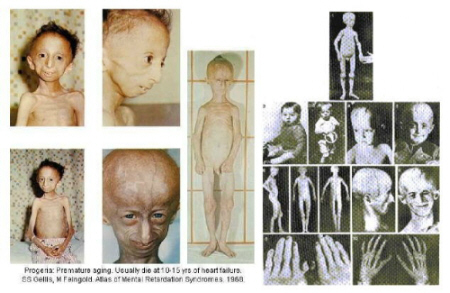
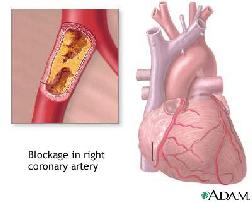 Dit alles lijdt uiteindelijk tot :
Dit alles lijdt uiteindelijk tot :
- Hart- en vaatziekten
- Aderverkalking
- Hartaanval(llen)
- Herseninfarcten & beroertes: bloeduistortingen in de hersenen
Alle symptomen uit het eerste en tweede levensjaar kloppen ook bij Michiel en Amber.
Collage Amber:

- Lengte – gewicht:
- De gemiddelde lengte bij Progeriakinderen van 12 jaar kan variëren tussen 96 cm. en 128 cm. Ze worden zelden groter dan 115 cm. Het gewicht ligt op 12-jarige leeftijd tussen 9,3 kg. tot 20,7 kg.
- Groei:
- Progeriakinderen groeien te traag. Vaak eet het kind ook slecht. Zowel de groei in gewicht als in lengte blijft achter maar het gewicht neemt het minst toe. Er treedt ook geen groeispurt op in de prepuberteit of de puberteit.
- Haar:
- Op enkele uitzonderingen na worden Progeria-kinderen geboren met normaal haar en een normale haarkleur. Het haar van deze kinderen groeit bijna niet, en op de leeftijd van zes maanden tot twee jaar valt het meestal uit. Tussen de twee en drie jaar is het haar bijna helemaal verdwenen. Er blijft vaak een klein beetje haar achter dat fijn, droog en donsachtig is. Bij hoge uitzondering heeft een Progeria-kind van 12 tot 15 jaar nog haar. Ook het haar van wenkbrauwen en oogleden verdwijnt, op sommige ooglidhaartjes aan de zijkant na. Het haar wordt licht van kleur.
- huidproblemen
- Een strakke harde huid (scleroderma) is een van de symptomen van Progeria die het eerst zichtbaar zijn, maar in de meeste gevallen is dit kenmerk van voorbijgaande aard. Bij sommige Progeria-kinderen werd scleroderma al op de eerste dag van hun leven geconstateerd, maar bij de meesten op een leeftijd tussen de anderhalf jaar en zes maanden. Ook later, op tweejarige leeftijd, kan dit voorkomen. De huid is dik, lichtelijk gezwollen en vertoont een drukpunt-oedeem (wanneer ingedrukt blijft een licht ‘deuk’ achter die langzaam wegtrekt). Soms is de huid roodachtig. Dit verschijnsel toont zich vooral op de onderbuik, boven de billen, op heupen en genitaliën, maar kan ook wijdverspreid zijn. Soms wordt de overgang tussen normale huid en sclerodermateuze huid gemarkeerd door een donkergekleurde zone. Scleroderma verdwijnt binnen zes maanden tot twee jaar tijd. Daarna wordt de huid dun, droog, atrofisch, minder elastisch en soms licht schilferig. De nek en het bovenste deel van de borstkas vertonen extra pigmentatie: kleine lichtbruine vlekjes. Later verschijnen deze vlekjes ook op de schedel en ledematen.
- Verlies van onderhuids vet en duidelijk zichtbare bloedvaten
- Verlies van onderhuids vet kan al optreden vanaf de leeftijd van zes maanden, maar wordt over het algemeen pas zichtbaar rond drie- tot vierjarige leeftijd. Dit is het eerst te zien aan de ledematen, vervolgens aan de borstkas en schedel en tenslotte in het gezicht. Tot die tijd hebben de kinderen vaak bolle blozende wangetjes. Het onderhuidse vetlaagje van buik en heupen verdwijnt het laatst. Het vet in de buik zelf verdwijnt echter niet, vandaar het vaak opgezette buikje van Progeriakinderen. Door de verdwijning van het onderhuidse vet en het dunner worden van de huid worden de aderen duidelijker zichtbaar. Omdat ook het gezichtsvet verdwijnt, lijken de ogen enigszins bol.
- Gebit:
- Daarnaast hebben veel kinderen een onregelmatig gebit. De tanden en kiezen hebben niet voldoende ruimte in de kaak om netjes naast elkaar te kunnen groeien. Tanden en kiezen komen laat door: bij de meeste kinderen komen de eerste tanden door tussen de 8 en 24 maanden. Tandbederf komt veel voor. Tanden poetsen is lastig in zo´n klein mondje, maar ook de kinderen die wel goed hun tanden kunnen poetsen hebben over het algemeen snel last van tandbederf.
- Hart en bloedvaten:
- Aanvankelijk hebben de kinderen geen problemen met het hart of de bloedvaten. Ook de bloeddruk is normaal in de eerste jaren. Vanaf vier jaar, meestal tussen zes en acht jaar, kan hartgeruis worden gehoord. Vanaf het zesde tot achtste jaar kunnen de kinderen kortademig worden, met een verhoogde bloeddruk. Op röntgenfoto´s is soms te zien dat het hart licht vergroot is. Angina Pectoris komt vaak voor, evenals arteriosclerose. De vasculaire problemen kunnen overal in het lichaam tot uiting komen: benauwdheid en ademhalingsproblemen komen voor en ook herseninfarcten worden door de dichtgeslibde aderen veroorzaakt. Sommige kinderen herstellen volledig, anderen houden klachten na een herseninfarct.
- Skelet en gewrichten
- Een van de eerste symptomen van Progeria is het heel laat of niet sluiten van de schedelnaden. Dit geldt vooral de voorste fontanel, die zelfs open kan blijven tot in de puberteit. Het schedeldak is grotendeels afhankelijk van de groei van de hersenen en omdat de hersenen bij Progeria gewoon groeien blijft het schedeldak normaal tot groot. Dit, samen met het botverlies van de gezichtsbeenderen en kleine kaak, is er de oorzaak van dat de schedel relatief gezien erg groot is ten opzichte van het gezicht. Door het kleiner worden van de kaak kunnen de tanden en kiezen schots en scheef komen te staan. In het begin zijn de spieren goed zichtbaar door het verdwijnen van het onderhuidse vet, de beentjes zien er gespierd uit. Later neemt ook de spiermassa af. De schouderbladen zijn goed zichtbaar, ze steken uit. Door artrose neemt de mobiliteit van de gewrichten steeds verder af, ook die in enkels, polsen, schouders en heup. Daardoor worden de gewrichten pijnlijk. Een verkorting van (een van) de nekspieren kan optreden en veel kinderen krijgen een enigszins bolle rug.
- mobiliteit
-
De beweeglijkheid van de gewrichten is normaal bij de geboorte. De knieën zijn meestal de eerste gewrichten waarin stijfheid optreedt, gevolgd door ellebogen en vingers. Dit gebeurt rond het tweede tot derde jaar. De stand van de benen is afwijkend doordat het bovenbeen naar buiten gedraaid is.
Daarom lopen de kinderen stijf, moeizaam en wijdbeens en met gebogen knieën (horse riding stance). In het begin zijn de spieren goed zichtbaar door het verdwijnen van het onderhuidse vet, de beentjes zien er gespierd uit. Later neemt ook de spiermassa af. De schouderbladen zijn prominent zichtbaar, ze steken uit. Door artrose neemt de mobiliteit van de gewrichten steeds verder af, ook die in enkels, polsen, schouders en heup. Daardoor worden de gewrichten pijnlijk. Een verkorting van (een van) de nekspieren kan optreden en veel kinderen krijgen een enigszins bolle rug. In laatste stadia kunnen de kinderen last hebben van artritis, als secundair verschijnsel. - andere symptomen
-
– Bijna alle progeria-patiëntjes hebben een hoog schril stemmetje.
– Een ander symptoom is een onregelmatig gebit. De tanden en kiezen hebben niet voldoende ruimte in de kaak om netjes naast elkaar te kunnen groeien. Tanden en kiezen komen laat door: bij de meeste patiëntjes komen de eerste tanden door tussen de 8 en 24 maanden. Tandbederf komt veel voor. Tanden poetsen is lastig in zo´n klein mondje, maar ook de kinderen die wel goed hun tanden kunnen poetsen hebben over het algemeen snel last van tandbederf. Er is hiervoor nog geen goede verklaring gevonden.
– Gehoorafwijkingen komen niet voor bij Progeria-kinderen. Ook op latere leeftijd blijft het gehoor redelijk goed functioneren. Ook staar komt op een zeer grote uitzondering na niet voor.
– Ook al hebben sommige Progeria-kinderen last van regelmatig terugkerende luchtweginfecties en oorontstekingen, onderzoek geeft geen afwijkingen aan op dit gebied.
– De genitaliën van Progeria-jongetjes worden in de literatuur omschreven als normaal, sommigen hebben een enigszins kleine penis. De zaadballen zijn normaal ingedaald. Door diverse onderzoekers is afwezigheid van spermavorming gemeld. Er zijn geen mannelijke Progeria-patiënten uit de geschiedenis bekend die vader zijn geworden. Vrouwelijke Progeria-patiënten hebben eveneens normale geslachtsorganen. Borstontwikkeling is echter afwezig. Ook is er geen oksel- of schaamhaar. Menstruatie treedt zelden op bij oudere Progeria-meisjes. In de literatuur wordt één meisje genoemd dat op 14-jarige leeftijd voor het eerst ging menstrueren, ze had een onregelmatige cyclus. In de literatuur wordt daarnaast nog een 32-jarige vrouw met niet-klassieke Progeria beschreven, die op 12-jarige leeftijd ging menstrueren en op 23-jarige leeftijd moeder werd van een gezond kind. Dit zijn echter uitzonderingen: over het algemeen komen Progeria-kinderen nooit in de puberteit en ontwikkelen ze geen geslachtshormonen. Wij kennen echter verschillende Europese progeriameisjes/jongvolwassenen die ongesteld worden met regelmatige cyclus.
– Van alle Progeria-kinderen uit de literatuur is slechts één 13-jarig meisje bekend dat kanker kreeg, dr. Hennekam kent een tweede kind met een vergelijkbare tumor.
In 62 wetenschappelijke rapporten uit het literatuuronderzoek van Dr. Hennekam, wordt de verstandelijke ontwikkeling van de Progeria-patiënten expliciet genoemd. Vier kinderen vertoonden een lichte achterstand, alle anderen werden als normaal beoordeeld. De intelligentie van Progeria-patiënten kan dan ook normaal worden genoemd. Van specifieke gedragsproblemen is geen sprake. De kinderen zijn vaak opmerkelijk alert, actief, optimistisch en vrolijk.
6. Erfelijkheid:
Er bestaat een theoretische, uiterst zeldzame mogelijkheid dat een Progeriakind een broertje of zusje krijgt met het Hutchinson-Gilford syndroom. Er zijn hierop twee uitzonderingen met betrekking tot eeneiige tweelingen:de Amerikaanse ééneiige tweeling (jongens) Chris en Chad (1978) en de Maleisische tweeling (meisjes) Lim Sook Khuen en Lim Sook Wen. De 4 kinderen zijn inmiddels overleden. Nu zijn er dan ook Michiel & Amber bekend. (Hieronder op foto) 

Bij een nieuwe zwangerschap kan een prenatale test uitsluitsel geven. Progeriapatiënten krijgen de kans niet om zich voort te planten. Ze sterven vaak te jong, en bovendien verhindert de verstoorde hormonale ontwikkeling dat de Progeriapatiënt vruchtbaar wordt. Als ze de mogelijkheid zouden hebben om zich voort te planten, zouden ze een grote kans hebben dat hun kinderen ook Progeria zouden hebben. In India is er een gezin met 5 Progeriakinderen, namelijk de kinderen Kahn. Zij lijden echter niet aan de klassieke vorm van Progeria maar aan een variante daarvan. Je ziet ook duidelijk de verschillen tussen alle kinderen met HGPS, (onze kinderen) en de kinderen Kahn :
- kinderen met klassieke Progeria (zoals onze kinderen) : schelle stem, krijgen nooit de baard in de keel, kaal vanaf 2 jaar, smal hoofd, spitse neus, hele kleine tanden en hartproblemen is doodsoorzaak nummer 1, lijken ook allemaal op elkaar.
- kinderen Kahn : één jongen had de baard in de keel en een zware stem, ze hadden nog allemaal haar (zelfs die van 15 en 19 jaar), ronde gezichten, grote brede tanden en zij hadden geen problemen met het hart.
-
8. Lichamelijke en geestelijke ontwikkeling :
Bij klassieke progeria vormt hartfalen de belangrijkste doodsoorzaak. Het levende bewijs daarvan is de 9-jarige Amerikaanse jongen Ory Barnett. Hij kreeg een hartaanval toen hij amper 5 jaar was. Ook al is hartfalen de belangrijkste doodsoorzaak bij kinderen en jongvolwassenen met Progeria, toch blijkt na grondig onderzoek dat de vijf Indische broers en zussen geen enkel hartprobleem hebben. “Dat is enerzijds goed nieuws omdat ze daaraan niet zullen sterven. Maar ze takelen wel even snel af. Hun borstkas dreigt in te stuiken en het beendergestel is te zwak aan het worden. We hebben absoluut geen idee welke toekomst deze kindjes te wachten staat”, zeggen artsen. Desalniettemin is het lot van de kinderen Kahn even erg als dat van bij ons.
7. X-Rays:
(left : normal person / right : Progeriachild)

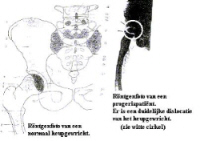

X-Rays : FALLER A., Het menselijke lichaam (bouw en functie), Oosthoek, Utrecht, 1975
De lichamelijke gevolgen van Progeria zijn vooralsnog niet te voorkomen. Progeria is nog niet te genezen, de medische wetenschap probeert tot nu toe symptomen te behandelen. Door vaatvernauwing is er al jong een sterk verhoogd risico op doorbloedingproblemen in de hersenen of een hartaanval. Om deze risico’s te verkleinen wordt er preventief een lage dagelijkse dosis aspirine voorgeschreven. Ondanks de vele overeenkomsten ontwikkelt de ziekte zich toch bij ieder kind anders. Door de algehele constitutie van het kind te versterken met bijvoorbeeld voedingssupplementen zal de levenskwaliteit verbeteren. De verstandelijke ontwikkeling van kinderen met Progeria verloopt normaal. Ze hebben geen achterstand op intellectueel gebied, op school zijn het vaak pientere en leergierige kinderen. Kinderen met Progeria ontwikkelen hun vaardigheden net zoals hun normale leeftijdsgenootjes. Alleen motorisch hebben de meeste kinderen een kleine achterstand. Michiel heeft ook problemen met de kleine motoriek : bv. hij schrijft niet graag omdat hij pijn in zijn vingers krijgt; van verven en kleuren krijgt hij krampen, e.d. Aangezien hij het alfabet kent, maakt hij gebruik van een speciaal voor hem op maat gemaakte computer. Het is echter geen spelcomputer. Zo raakt hij ook niet achter in de klas en kan hij het tempo volgen. Ook zijn schooltaken of huiswerk wordt op deze computer door hem gemaakt. 
Toch kunnen bijna alle kinderen met Progeria ondanks alles met hart en ziel leven: ze beginnen vol vreugde aan elke nieuwe dag, en genieten van de mooie dingen die het leven te bieden heeft. Ze hebben een hele klare kijk op het leven, zitten vol levensvreugde en dankzij hun open, vriendelijke en levenslustige aard worden ze algauw aanvaard door de klasgenootjes. Door hun ongewone voorkomen worden kinderen met Progeria buiten hun vertrouwde leefomgeving vaak aangestaard; een situatie waarmee zij, en hun ouders, moeten leren omgaan. Ze blijken vaak ad rem en humoristisch te kunnen reageren op de soms tactloze opmerkingen. Misschien daarom vinden ze op school toch meestal snel vriendjes en vriendinnetjes.
9. Emotionele proces:
De diagnose Progeria is voor familieleden een grote schok. Door hun ongewone voorkomen worden kinderen met Progeria buiten hun vertrouwde leefomgeving vaak aangestaard. Het gebeurt dat voorbijgangers op straat confronterende vragen stellen. Onhandige reacties van vreemden kunnen erg kwetsend overkomen. Dat zijn pijnlijke situaties waarmee het kind en de ouders moeten leren omgaan.Vaak gaan er dan ook jaren voorbij voordat de diagnose wordt geaccepteerd en het ‘leven met Progeria’ als positief kan worden ervaren. Het is een moeilijke opgave om te leren omgaan met het perspectief, de lichamelijke gevolgen van de ziekte en de soms botte reacties uit de omgeving. Maar het belangrijkste is natuurlijk dat het kind volledig wordt geaccepteerd en voelt dat er van hem of haar gehouden wordt. Onvoorwaardelijke liefde is ook voor deze kinderen onmisbaar, hij/zij moet zoveel mogelijk gewoon kind kunnen zijn. Ook voor het kind kan het moeilijk te aanvaarden zijn om deze ziekte te hebben en de gevolgen en pijn ervan te dragen.
De meeste (oudere) kinderen overlijden allemaal wetende wat er aan de hand is. Ze beseffen het heel goed. Deze kinderen en hun ouders blijven vaak zitten met vele vragen die niet kunnen worden beantwoord. Na het stellen van de diagnose ligt er voor de ouders een moeilijke opgave: hoe kun je deze ziekte accepteren? Kun je je ondanks het verdriet toch instellen op de ontwikkelingsmogelijkheden van je kind? Dat is de eerste en de moeilijkste opgave voor de familie nadat de diagnose Progeria is gesteld. Een pijnlijk en zwaar proces dat veel tijd vraagt. Als ouders moeten we ons erop richten dat het kind zich lichamelijk anders zal ontwikkelen. Door de definitieve diagnose worden we gedwongen onze eigen voorstellingen en verwachtingen van het leven te herzien.
Als vader of moeder worstel je met vragen over de problemen die in de loop van de tijd op je af zullen komen. En alle vragen die het kind je zelf zal stellen zul je ook zo open mogelijk moeten beantwoorden op een manier die past bij de leeftijd van het kind. Daarmee leer je het kind zichzelf te accepteren zoals hij is. Sommige ouders isoleren zich, kunnen niet praten over dit leed, andere ouders hebben juist nood om erover te praten. Het is heel belangrijk dat een ouderpaar elkaar hierin goed aanvoelt. Of het brengt je dichter bij elkaar, of het drijft je uit elkaar. Terwijl er in sommige gezinnen ook nog broertjes en zusjes zijn die ook een moeilijk emotioneel proces moeten doorstaan.
Ook is het belangrijk dat het kind gevoel van zelfstandigheid heeft en het gevoel van eigenwaarde. Michiel wil bv. alles zelfstandig doen, wil niet geholpen worden waardoor er bij ons thuis de nodige aanpassingen gebeurd zijn, met name : de deurklinken en lichtknoppen verlaagd, een bijkomende trapleuning geplaatst aangepast aan zijn lengte, de badkamer volledig aangepast : een wastafel geplaatst op zijn lengte, een opstapje aan het bad en toilet, doucheknop op zijn lengte voorzien, en dergelijke meer.
Aanpassingen in huis: 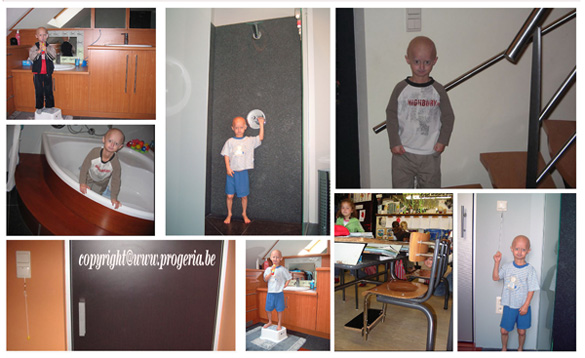 De verwachtingen veranderen. Bij Progeria kunnen verwachtingen over “later als ik groot ben” vaak niet meer. Het kind zal ook steeds minder kunnen. Verwachtingen op lange termijn zijn er bijna niet meer en ouders willen ook niet meer denken op lange termijn. Ze willen genieten van elke dag. De ouders leven van dag tot dag en zien wel wat er wel en niet kan en gebeurt. Michiel kan helemaal alleen zelfstandig zwemmen. Dat is een hele stap vooruit en wij zijn natuurlijk heel trots op deze prestatie. Doordat hij geen onderhuids vet heeft, koelt hij wel vlug af in het water (zijn lippen, handen kleuren dan blauw) waardoor wij hem een wetsuit gekocht hebben en hij daardoor langer in het water kan vertoeven.
De verwachtingen veranderen. Bij Progeria kunnen verwachtingen over “later als ik groot ben” vaak niet meer. Het kind zal ook steeds minder kunnen. Verwachtingen op lange termijn zijn er bijna niet meer en ouders willen ook niet meer denken op lange termijn. Ze willen genieten van elke dag. De ouders leven van dag tot dag en zien wel wat er wel en niet kan en gebeurt. Michiel kan helemaal alleen zelfstandig zwemmen. Dat is een hele stap vooruit en wij zijn natuurlijk heel trots op deze prestatie. Doordat hij geen onderhuids vet heeft, koelt hij wel vlug af in het water (zijn lippen, handen kleuren dan blauw) waardoor wij hem een wetsuit gekocht hebben en hij daardoor langer in het water kan vertoeven.
Alle stapjes vooruit is voor ons telkens een mijlpaal waar wij alleen maar trots op kunnen zijn Bij de ziekte Progeria blijft de verzorging ook altijd. Gedurende een tijdje neemt deze misschien wat af maar neemt weer toe naarmate de kinderen meer problemen krijgen. De grootste hulp voor een kind met Progeria zijn natuurlijk de ouders die hun kind volledig steunen. Maar bij wie kunnen de ouders terecht : Wanneer jij als ouder het moeilijk hebt, wie staat er dan voor je klaar om je op te vangen ? Is er iemand waarop jij eens terug kan vallen ? Zijn er mensen die met je meedenken, die bereid zijn je mee te helpen, waar je terecht kunt met je vragen ? Het is dus een zwaar, lang en onzeker proces voor de ouders. Hierbij hebben ze de steun van anderen nodig (partner, familie, vrienden, lotgenoten).
Ze ondergaan echt een heel moeilijk stadium in hun leven, maar ze hebben bijna geen andere keus dan ermee te leren leven. De meeste ouders aanvaarden het dan ook wel na een tijd, uiteraard mede door de hulp van anderen. Ze moeten het uiteindelijk een plaatsje in hun hart geven want anders is het ondraaglijk. En natuurlijk hebben ze nog altijd hun goede en kwade momenten. Als ze het al een keertje moeilijk hebben, moeten ze ook tegen elkaar kunnen zeggen dat het die dag niet gaat en ze mogen zich ook eens afzonderen om hun traantjes te laten vloeien (met of zonder elkaar). De ouders moeten zich niet steeds sterk houden want er moet ook de ruimte zijn om het verdriet even de bovenhand te laten. Ook bij Godelieve en Wim was dit het geval. Ze konden nergens anders terecht dan bij familie / vrienden en nadien bij Marjet Stamsnijder, de voorzitster van de Progeria Family Circle die hen heeft voorgesteld aan andere gezinnen met een kind die lijdt aan Progeria en die hierdoor hetzelfde doorstaan.
Het is dus zeker nodig beroep te doen op anderen, want je wordt psychologisch niet opgevangen na zo’n diagnose. Vanuit het ziekenhuis wordt je door de professor of dokter de diagnose medegedeeld en dat is het. Je ziet maar hoe je het verwerkt. Er is niemand naar wie je doorverwezen wordt. De psychologische hulp die je dan toch heel erg nodig hebt, is zeer mager en beperkt. Aangezien je toch die enorme behoefte hebt aan steun, zoek je die toch ergens anders : bij je partner, je zus, je beste vriend of vriendin, familie. Je zoekt iemand waarbij je je geborgen voelt en waarbij je gemengde gevoelens en je verdriet kunt uiten. Het feit is er dat éénmaal de diagnose hardop gezegd wordt en je weet dat je kind vroegtijdig zal komen te overlijden, je met verschillende gevoelens geconfronteerd wordt : voornamelijk verdriet, angst, ontkenning, hulpeloosheid, verwarring, krachteloosheid, woede, boosheid, wanhoop, depressiviteit, ….. misschien wel schuldgevoel. Ouders van een Progeriakind maken een fase door van diepe, onvermijdelijke rouw.
Door de bekendheid van Progeria hoeven de ouders echter niet meer bang over straat te lopen. Voorbijgangers kijken hen niet meer langer na alsof hun kind een enge ziekte heeft. Overal ter wereld komt er nu meer bekendheid over de ziekte. Eerst dachten voorbijgangers bijvoorbeeld dat het besmettelijk zou kunnen zijn en liepen ze er ook met grote bogen omheen. Tegenwoordig weet men beter. De kinderen mogen weer met deze kinderen spelen en de ouders worden bemoedigend toegesproken. Ook hiervan is Michiel weer een mooi voorbeeld. Hij is door zijn bekendheid Limburger van het jaar 2003 geworden. Men zou hem niet hebben verkozen als ze hem eng zouden hebben gevonden; nee, ze bewonderen hem.
Ze zoeken steun bij andere ouders, wisselen ervaringen uit, enz. Het kind wordt soms geremd in zijn/haar sociale ontwikkeling omdat de conditie en toestand het gewoon niet toelaat. Sociaal contact is echter heel belangrijk voor een kind, ze moeten zich vrij kunnen uiten en ontwikkelen. Tenslotte hebben deze kinderen dezelfde behoeften als die van een gewoon kind, ze spelen ook graag, willen ook bepaalde zaken doen die andere kinderen doen, bv: Michiel houdt erg veel van voetbal en is ook aangesloten bij een voetbalploeg alhoewel hij natuurlijk niet de conditie heeft om een hele match te spelen. Op de speelplaats zie je hem gewoon meespelen met de andere kinderen en hij weet zelf zijn grens liggen (hij weet hoe ver hij kan gaan) hetgeen ook heel belangrijk is. Als hij niet meer kan, zegt hij gewoon dat hij even moet rusten en dat hij later weer terug mee zal doen. De kinderen begrijpen dat ook en houden daar ook rekening mee.
Doordat ze er ‘anders ‘ uitzien dan andere mensen worden ze vaak aangestaard of krijgen ze soms negatieve en kwetsende opmerkingen. De mensen ontwijken liever deze kinderen dan ze een lach te gunnen, en dat is juist wat de ouders willen vermijden door Progeria in de kijker te brengen en de mensen te laten kennis maken met hun situatie en met die van alle andere gezinnen. Dit was bij Michiel ook zo; de mensen keken hem raar aan en lieten hun kinderen niet in de buurt komen omdat ze bang waren voor Michiel of gewoonweg voor besmetting. Overal waar wij kwamen, staarde men hem aan (zelfs met open mond), draaiden de mensen zich nog eens om hem na te staren, waren ze aan het fluisteren over hem en aan het wijzen met de vinger, e.d. En vooral dat kwetste Godelieve heel erg. De aanvaarding in de maatschappij loopt dus niet van een leien dakje.
Daarom dat Godelieve en Wim besloten hebben daaraan iets te doen, en de media te gebruiken als hun middel om Michiel (en alle andere kinderen) te laten aanvaarden. Zodat ze meer respect en begrip krijgen. Dat is hen ook gelukt. Toen de mensen wisten wat er scheelde, toonden ze erg veel begrip voor Michiel en boden zelfs hulp aan. Ze worden nu goed opgevangen door hun omgeving en zo zijn de kinderen ook vrij om zich volledig te ontwikkelen en te uiten. Deze kinderen hebben allemaal een aangenaam, vrolijk en sociaal karakter en zijn gemakkelijk in hun omgang met anderen. Sinds de artikels in de kranten en de reportages op televisie is ons leven er toch een stukje mooier op geworden. Alleen al het feit dat wij op straat kunnen lopen en de mensen ons niet meer nastaren maar hem toejuichen, laat ook het zonnetje in ons hart extra schijnen. Daar hadden wij wel nood aan : begrip, aanvaarding van het “anders-zijn” van ons zoontje en sympathie. Contact met andere families met lotgenootjes : Héél belangrijk is het contact met andere Progeria-families.
Het contact met andere kinderen en hun ouders blijkt altijd veel emotionele ondersteuning te geven, ouders vinden antwoord op hun vragen en krijgen waar mogelijk hulp. Kinderen met Progeria bouwen in de ontmoeting met elkaar veel zelfvertrouwen en levenslust op.

Tijdens een reünie kan men andere families en artsen om raad en advies vragen, specifieke problemen bespreken, en betrokken families vinden bij elkaar de informatie en hulp die ze zo hard nodig hebben bij hun moeilijke levenstaak. Onderling contact is een bron voor meer levenskwaliteit. Ik moet eerlijk toegeven dat de bij onze allereerste reünie, de confrontatie met vooral de oudere kinderen, heel emotioneel was. Ik huilde voortdurend. De ouders vertelden elk hun verhaal. Het ene nog erger dan het andere. Kinderen die gepest werden op school, in de vuilbak gesmeten omdat ze “anders uitzien”, die al hartklachten hadden maar desondanks toch ook het mooie verhaal van Burçu die vertelde dat doordat zij een kind is met een zeldzame ziekte, ze de mogelijkheid heeft gehad om contact te hebben met een Turkse zanger, haar idool. Had zij geen Progeria gehad, dan had ze die man misschien nooit ontmoet. Je ziet ook de veroudering die zeer merkbaar is en dan dringt het tot je door dat je eigen kind er ook zo zal gaan uitzien.
Ik schrok. Aan hen zie je het toekomstbeeld voor ogen zoals Michiel uiteindelijk eruit gaat zien. Enorm moeilijk had ik het. Je stelt je duizend en één vragen daar : Zal hij ook zo een last hebben van onderhuidse pijnen ? Zal hij net zoals Menekçe op een tijdstip komen dat hij zelf niet meer kan gaan zitten of terug rechtstaan ? Zal hij al vroegtijdig last hebben van zijn hart ? Van diabetes ? Van tandpijnen ? Slapeloze nachten en als je dan uiteindelijk slaapt, achterhalen alle verhalen je, de gezichtjes van de kinderen komen in je dromen terug en ook het verdriet van de ouders. Je hoort een kind van 16 zeggen dat ze meer pijn heeft gehad in haar leven dan dat ze gelukkig geweest is; dat te horen doet heel veel pijn. Maar het is de werkelijkheid; de realiteit. Tijdens die eerste meeting met alle kinderen zat ik emotioneel heel erg in de knoop maar desondanks had ik het niet willen missen. Je voelt je thuiskomen, je voelt je geen rariteit meer en de kinderen onderling weten dat ze niet de enigen zijn die geen haar hebben en waarvan de aders zichtbaar zijn op het hoofd. Ze voelen zich verbonden met elkaar.
Ze lijken ook zo fel op elkaar. De daarop volgende weken had ik het enorm moeilijk. Ik zat emotioneel in de knoop. Tot ik het een plaatsje kon geven. Ondanks de eerste emotionele confrontatie met de andere kinderen, keken we enorm uit naar de volgende ontmoeting en ondertussen hebben we elkaar al meermaals ontmoet.
De allereerste angst ebde volledig weg. Als we elkaar tijdens een meeting terugzien, zijn wij allemaal één familie. Alle families hebben een band met elkaar gesmeed voor het leven ondanks de verschillende nationaliteiten, achtergrond en vooral de taal. Wij lachen, huilen, praten samen maar genieten vooral allemaal voor eventjes van een onbezorgde tijd.
Al deze kinderen hebben een heel bijzondere plaats in ons hart ingenomen. Ze beschikken allemaal over de bijzondere gave om iemand op te monteren, ze zijn heel spontaan, sociaal, goedlachs en levenslustig en wij kijken al lang niet meer naar hun uiterlijk omdat ze zo een bijzonder innerlijk hebben. 
Wij hopen met onze ervaring een steun te kunnen zijn voor Liesbeth en Gerry (Toon*) en voor Franky en Martine (Mats*).
Tussen de drie gezinnen is er contact en dat is héél belangrijk en wij hebben er allemaal ook behoefte aan.
Door onze kinderen is er een speciale band voor het leven. Zij kunnen bij ons terecht voor hulp, informatie, begeleiding zoals ook wij raad kunnen vragen aan de andere families met een ouder kind die lijdt aan Progeria.
Wij kunnen bij elkaar ervaringen uitwisselen en vooral een warme vriendschapsband smeden.
10. Hulp :
Gezien het ziekteproces bij Progeria moet steeds worden afgewogen in hoeverre medische hulp noodzakelijk is. Het is niet goed om het kind te belasten met overmatige zorg en medicatie. Ouders en artsen moeten steeds afwegen in hoeverre de belasting van een behandeling opweegt tegen de vermoedde effecten.
- Experimentele behandeling met menselijke groeihormonen hebben kinderen met Progeria bijvoorbeeld niet geholpen. Deze behandeling wordt ook niet meer toegepast, omdat men niet kan uitsluiten dat het toedienen van menselijk groeihormoon het verouderingsproces bij Progeria versnelt.
- In het buitenland heeft ook het toedienen van dierlijk groeihormoon (samengesteld uit embryonaal dierlijk weefsel) niet tot genezing geleid. De ouders van kinderen en jongvolwassenen met Progeria die met deze celtherapie zijn behandeld, vonden wel dat de kinderen er constitutioneel veel baat bij hadden. Deze medicatie is echter zeer omstreden, en in Nederland dan ook niet toegestaan.
- Hartchirurgie (in het verleden vaak toegepast op kinderen met Progeria) heeft vaak maar een beperkt effect, terwijl zo’n operatie voor een progeriakind grote risico’s met zich mee brengt.
Sommige ouders hebben positieve ervaringen met behandelingen en therapieën:
- Met het oog op cardiovasculaire klachten wordt er door artsen meestal geadviseerd om preventief om de dag (soms dagelijks) een lage dosering Aspirine te geven. De dosering is afhankelijk van de constitutie van het kind: 3-5 mg per kilogram lichaamsgewicht. Stop met aspirine wanneer het kind veel blauwe plekken krijgt of een wondje te lang blijft doorbloeden, en geef minstens een week geen aspirine voordat er een chirurgische ingreep (ook bij de tandarts) plaats moet vinden. Wees ook voorzichtig wanneer er waterpokken heerst: raadpleeg in alle gevallen uw arts. Verder zijn er nauwelijks bijwerkingen bekend, slechts een enkel kind kreeg hiervan wat last van zijn maag.
- Zachtaardige fysiotherapie en veel aangepaste lichamelijke oefening, zoals zwemmen in warm water. Dit werkt de gewrichtsverstijving tegen. De spieren worden daardoor versterkt en de doorbloeding wordt bevorderd.
- Michiel wordt 2 x per week door de kinesist gemasseerd teneinde zijn spieren en gewrichten soepel te houden. Deze heeft een speciale behandeling voor Michiel uitgewerkt. Die moet de pijn in Michiels heupgewrichten verzachten.
- Aan de slechte eters wordt soms een voedingssupplement gegeven wanneer de caloriebehoefte van het kind groter is dan wat het bij de maaltijden naar binnen kan krijgen. De kinderarts of diëtiste kan als aanvulling op de gewone dagelijkse maaltijden bijvoorbeeld Pediasure of Ensure voorschrijven.
- Belangrijk is een goede verzorging van de melktandjes. Tandenwisseling vindt meestal maar gedeeltelijk plaats, hoewel de tweede tanden in aanleg wel aanwezig zijn. In de kleine kaak is meestal weinig ruimte voor het gebit, toch moet men voorzichtig zijn met ingrijpende gebitsregulatie.
- Aanschaf van noodzakelijke hulpmiddelen. Sommige kinderen willen graag een pruik wanneer ze naar school gaan. Voor kinderen met gewrichtsproblemen of een verminderde hartfunctie maakt een aangepaste fiets het vaak mogelijk om toch zelfstandig naar school te gaan.
- Medicinale badoliën en bodylotions kunnen helpen om jeuk te verzachten en ze gaan droogte tegen van de dunne huid.
- Aanpassingen in huis zoals bijvoorbeeld een bereikbare deurklink of een lage wastafel bevorderen de zelfstandigheid van het kind, en dragen bij aan het gevoel van eigenwaarde.
- Sommige kinderen willen na verloop van tijd graag een pruik, jongens dragen dan bij voorkeur een petje. Voor kinderen met gewrichtsproblemen of een verminderde hartfunctie maakt een aangepaste fiets het vaak mogelijk om zelfstandig naar school te blijven gaan.
11. Behandeling:
Hoewel de ziekte Progeria niet kan genezen worden, hebben ze wel een medicijn gevonden om de gevolgen ervan tegen te gaan. Op chromosoom 1 ligt het LMNA gen. Dit gen bevat de code voor de aanmaak van 2 eiwitten vl. het prelamin A en het lamin C. In normale omstandigheden wordt het prelamin A dan verder ‘verwerkt’ tot lamin A.
Die ‘verwerking’ gebeurt in verschillende stappen : eerst is er inwerking door een enzyme genaamd farnesyltransferase (hierdoor wordt er een ‘farnesylgroep’ (Een farnesylgroep is een scheikundige groep die bestaat uit een opeenvolging van koolstofatomen (=een 15-tal). Deze farnesylgroep wordt op het prelamin A geplaatst en is één van de stappen in de omvorming van prelamin A naar het afgewerkte Lamine A.) op de prelamin A geplaatst), daarna gebeurt er een ‘carboxymethylatie’ (op het einde van het eiwit wordt het -COOH stukje omgezet naar -COCH3), uiteindelijk gebeurt er een ‘endoproteolysis’ (afsplitsing van een groot stuk van het eiwit) en het lamin A is afgewerkt. Het lamin A en het lamin C, samen met lamin B vormen dan een ‘multiproteinen’ complex aan de binnenrand van het kernmembraan waar ze een zeer belangrijke rol spelen bij de uitwisseling van gegevens tussen de kern en het cytoplasma van de cel.
Bij kinderen met progeria is er een foutje in het LMNA gen. Hierdoor wordt een afwijkend ‘prelamin A’ gevormd. De eerste verwerking, nl. farnesylatie (een farnesylgroep op het eiwit plaatsen) is wel mogelijk maar de endoproteolysis kan op dit afwijkend eiwit niet gebeuren. Hierdoor ontstaat een eiwit, genoemd het ‘progerin’, dat langer is dan het normale lamin A en niet kan ingebouwd worden in het ‘multiproteinen’ complex van de kernwand maar neerslaat en opstapelt in de kern. Dit veroorzaakt een storing in de normale werking van de kern, en dus van de gehele cel. De medicatie welke nu uitgetest wordt gaat de werking van het farnesyltransferase tegen (=farnesyltransferase inhibitoren of FTI ‘s). Hierdoor wordt er geen extra lang progerin gevormd en is er dus geen opstapeling van dit eiwit in de kern van de cel.
Een farnesylgroep is een scheikundige verbinding dat bestaat uit meerdere atomen. Deze uitleg dient om te helpen in het begrijpen van het ingewikkeld scheikundig proces dat aan de basis ligt van het ontwikkelen van nieuwe medicatie voor progeria. Farnesyltransferase remmers : Lonafarnib Onderzoek uit 2007 toonde aan dat de opbouw van onvolledig Lamine A voorkomen kan worden met farnesyl transferase inhibitoren (FTI’s). Het LMNA gen levert bij gezonde mensen de code waarmee het eiwit prelamine A wordt gemaakt, waaruit vervolgens lamine A wordt gevormd. De mutatie bij kinderen met progeria leidt echter tot de productie van een abnormale vorm van prelamine A, men noemt dit progerin. FTI’s zijn een geneesmiddel waarmee de toxische werking van progerin wellicht kan worden tegengegaan.
De behandeling van muizen met progeria gaf hoopvolle resultaten. Men onderzoekt nu in Boston of farnesyltransferase remmers (FIT’s) een mogelijke behandeling kan bieden voor kinderen met HGPS. In 2007 is een trial gestart, waarbij kinderen met progeria met FTI’s, in de handel als het medicijn Lonafarnib® worden behandeld. De opbouw van prelamine A wordt hiermee geblokkeerd, en de verwachting is dat deze kinderen beter zullen functioneren wanneer het progerin in de celkern ontbreekt.
Zie ook:Clinical Research News January 2008 A) First-Ever Progeria Clinical Drug Trial in Boston : Een trial met deze therapie is gestart door het National Institute of Health door een Engels medisch team onder leiding van dokter Mark Kieran en Dr. Leslie gordon op 7 mei 2007 bij een groep van 28 kinderen met Progeria. De resultaten zullen in 2009 worden gepubliceerd. De kinderen komen uit 16 verschillende landen (Argentinië, België, Canada, Denemarken, Engeland, India, Israël, Italië, Japan, Mexico, Pakistan, Polen, Portugal, Romenië, Verenigde Staten en Venezuela) en de leeftijd varieert tussen 3 jaar en 15 jaar.
Per 4 maanden reizen telkens 2 kinderen naar Boston voor een hele week en dit gedurende een periode van 2 jaar. Dit onderzoek naar de werkzaamheid en de effecten van FTI’s (farnesyltransferaseremmers) is dus nog in de onderzoeksfase. Doordat de werkzaamheid van FTI’s in Progeriamuizen effectief bleek, wordt dit medicijn onder strikte begeleiding aan 28 Progeriapatiëntjes gegeven. De trial wordt in Boston uitgevoerd Het eerste verblijf in het Children’s Hospital in Boston duurt 1 week. In deze week worden heel uitgebreide testen gedaan (röntgenfoto’s, hartonderzoek, gewicht, lengte, bloeddruk, enz.) en wordt er gestart met de kuur. De dokters in hun thuisland dienen de eerste maand elke week eenmaal bloed te trekken van het kind en samen met een periodiek gezondheidsrapport op te sturen naar het medische team in Boston en daarna 1 maal per maand en dit gedurende een periode van 2 jaar.
Om de 4 maanden zal men terug moeten gaan naar Boston, voor verdere onderzoeken, eventuele aanpassing van de dosis en men krijgt dan een nieuwe voorraad mee voor de eerstvolgende 4 maanden. De medicatie geschiedt dagelijks in pilvorm, namelijk 2 x 2 pillen, ’s morgens en ’s avonds met een tussenpauze van 12 uur. Sinds Michiel gestart is met zijn medicatie is hij 5 cm. gegroeid, hij krijgt lichte donshaar, zijn aders zijn dikker en zijn huid is steviger. Hij heeft als bijwerking enkel diarree. Hier zie je een foto van een normale cel, een progeriacel en hoe de progeriacel eruit gaat zien na behandeling met de FTI.
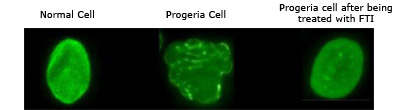
B) Nieuwe hoop voor kinderen met Progeria
1 Juli 2008 / Vasten® en Zometa® Nadat vijf jaar geleden werd ontdekt welk gen is betrokken bij het ontstaan van de ziekte Progeria, is er naarstig gezocht naar een therapie die voortijdige veroudering kan tegengaan. Een team onderzoekers onder leiding van Nicolas Lévy in Marseille (Frankrijk) en Carlos López-Otín in Orviedo (Spanje) publiceerde vandaag opzienbarende resultaten in het vakblad Nature Medicine.Progeria wordt veroorzaakt door een onvolledig opgebouwd eiwit (Lamine A) in de celkern. Het lichaam van progeria patiëntjes kan dit eiwit, dat o.a. de celkernwand moet verstevigen, niet volledig opbouwen. Er blijft bij hen een vetzuur in de celkern achter, dat in normale cellen wordt afgebroken bij de opbouw van Lamine A. Er ontstaat een stapeling van vettig, verkeerd opgebouwd eiwit (men is dit progerin gaan noemen), wat grote problemen veroorzaakt.
De cel verliest zijn stevigheid en de functie wordt aangetast. Hierdoor blijft DNA-herstel achterwege. Mede hierdoor groeien kinderen met progeria niet goed, verliezen zij hun haar en onderhuids vet, en krijgen zij last van botontkalking en hart- en vaatproblemen.
Dit beperkt de levensverwachting sterk.  In de hoop de toxiciteit van het onvolledig opgebouwde eiwit tegen te kunnen gaan, heeft het Frans-Spaanse team opnieuw onderzocht op welke manier het Lamine A zich aan de celkernwand vasthecht.
In de hoop de toxiciteit van het onvolledig opgebouwde eiwit tegen te kunnen gaan, heeft het Frans-Spaanse team opnieuw onderzocht op welke manier het Lamine A zich aan de celkernwand vasthecht.
Er zijn namelijk medicijnen die fasen van deze eiwitopbouw kunnen remmen.
Eerder onderzoek toonde ook al aan dat de opbouw van onvolledig Lamine A voorkomen kan worden met farnesyl transferase remmers (FTI’s).
Met FTI’s wordt de eiwitopbouw geblokkeerd.
Dr. Carlos López-Otín onderzocht dit nader door de Lamine-A opbouw bij zijn progeria muismodellen te blokkeren. Hij constateerde dat wanneer de opbouw geblokkeerd wordt, het lichaam van muizen toch een nieuwe weg zoekt om dit eiwit te vormen. Via een andere weg ontstaat er desondanks toch stapeling van vetten in de celkern. Dr. Lévy en Dr. López-Otín ontwikkelden daarom een nieuwe behandeling voor een aantal vormen van Progeria, met name voor het Hutchinson-Gilford Progeria syndroom (HGPS).
Muizen met dezelfde genetische structuur als kinderen met HGPS kregen statines. Vasten®, een statine die veel wordt toegepast bij hart- en vaatziekten. Daarnaast gaven zij aminobifosfonaat (Zométa®). Bisfosfonaten worden gebruikt voor de behandeling van botontkalking. De combinatie van deze twee middelen blijkt de gezondheid van progeria-muizen te bevorderen. Statines en aminobifosfonaat kunnen stapeling van vetzuur in de celkern voorkomen. Daarmee worden de gevolgen van de ziekte tegengegaan. Hoewel de ziekte Progeria nog steeds niet genezen kan worden, ontwikkelden de muizen zich met deze medicatie beter en steeg de levensverwachting met 50%.  De volgende stap is dat vijftien Europese kinderen deze therapie aangeboden krijgen in het kader van een klinische studie. Dit klinisch onderzoek wordt financieel gesteund door de Franse Téléthon, en door het AFM (Association Française contre les Myopathies). Deze klinische studie zal de komende drie jaar plaatsvinden in het Hôpital d’Enfants de la Timone in Marseille. Het doel is om de voortgang van de ziekte te vertragen en naar wij hopen zal deze medicatie ook de levensverwachting van de getroffen kinderen verlengen. De publicatie in Nature Medicine: http://www.nature.com/nm/journal/vaop/ncurrent/abs/nm1786.html
De volgende stap is dat vijftien Europese kinderen deze therapie aangeboden krijgen in het kader van een klinische studie. Dit klinisch onderzoek wordt financieel gesteund door de Franse Téléthon, en door het AFM (Association Française contre les Myopathies). Deze klinische studie zal de komende drie jaar plaatsvinden in het Hôpital d’Enfants de la Timone in Marseille. Het doel is om de voortgang van de ziekte te vertragen en naar wij hopen zal deze medicatie ook de levensverwachting van de getroffen kinderen verlengen. De publicatie in Nature Medicine: http://www.nature.com/nm/journal/vaop/ncurrent/abs/nm1786.html
C) Boston : @ Progeria Research Foundation Augustus 2009 :
Aankondiging van de “Progeria Triple Drug Trial”
 De Progeria Research Foundation en het Children’s Hospital Boston zijn wederom de samenwerking aangegaan m.b.t. een tweede klinisch experiment voor kinderen met Progeria. In dit spannende en langer durende experiment zullen 45 kinderen uit 19 landen deelnemen. Onderzoekers hebben twee extra medicijnen geïdentificeerd die, wanneer ze gebruikt worden in combinatie met het huidige FTI-medicijn, een efficiëntere behandeling voor kinderen met Progeria zou kunnen verstrekken dan enkel met het FTI-medicijn. Progeria wordt veroorzaakt door een abnormale proteïne genoemd progerin. Het Progeria-onderzoeksteam van het Children’s Hospital Boston zal twee medicijnen toevoegen, genoemd “Pravastatin“ en “Zoledronate”, aan de huidige behandeling met FTI’s. De drie medicijnen zullen zich gaandeweg richten tot de productie van het ziekteveroorzakende progerin. In een opzienbare laboratoriumstudie, voorgeleid door Dr. Carlos Lopez-Otin van Spanje op de 2007 Progeria Research Foundation Scientific Workshop, verbeterden de twee nieuwe medicijnen de aandoening in progeriacellen en steeg de levensduur van muizen met Progeria.
De Progeria Research Foundation en het Children’s Hospital Boston zijn wederom de samenwerking aangegaan m.b.t. een tweede klinisch experiment voor kinderen met Progeria. In dit spannende en langer durende experiment zullen 45 kinderen uit 19 landen deelnemen. Onderzoekers hebben twee extra medicijnen geïdentificeerd die, wanneer ze gebruikt worden in combinatie met het huidige FTI-medicijn, een efficiëntere behandeling voor kinderen met Progeria zou kunnen verstrekken dan enkel met het FTI-medicijn. Progeria wordt veroorzaakt door een abnormale proteïne genoemd progerin. Het Progeria-onderzoeksteam van het Children’s Hospital Boston zal twee medicijnen toevoegen, genoemd “Pravastatin“ en “Zoledronate”, aan de huidige behandeling met FTI’s. De drie medicijnen zullen zich gaandeweg richten tot de productie van het ziekteveroorzakende progerin. In een opzienbare laboratoriumstudie, voorgeleid door Dr. Carlos Lopez-Otin van Spanje op de 2007 Progeria Research Foundation Scientific Workshop, verbeterden de twee nieuwe medicijnen de aandoening in progeriacellen en steeg de levensduur van muizen met Progeria.
Doel: Als de drie drugs, die in deze proef worden toegediend, effectief de farnesylgroep kunnen blokkeren, dan kan het progerin worden “verlamd” en progeria “verbeterd”. Wij hopen dat de medicijnen zullen samenwerken, om elkaar aan te vullen zodat de progerinprotëine meer beïnvloed zal worden door de combinatie van de drie medicijnen dan enkel het gebruik van één medicijn alleen.
Wie zal deelnemen in dit driedubbel experiment? Het haalbaarheidsexperiment: Het team heeft al een miniproef voor 5 kinderen met Progeria geleid. Dit haalbaarheidsexperiment dat één maand duurde, zou moeten vaststellen of de combinatie van de drie medicijnen goed zou worden verdragen, voorafgaand aan een groter internationaal experiment. De bijwerkingen waren verdraaglijk/aanvaardbaar en het team heeft zich volop gestort op het groter doeltreffendheidsexperiment.
Het doelbaarheidsexperiment: Wij voorzien dat 45 kinderen uit 19 verschillende landen, die 11 verschillende talen spreken, zich zullen inschrijven voor deze experimentele behandeling. Dit omvat kinderen die al deelnamen in de eerste FTI-behandeling, de 5 kinderen die deelnamen aan het haalbaarheidsexperiment, en de kinderen die destijds te jong waren om deel te nemen aan de eerste behandeling of kinderen die wij nog ontdekt hebben gedurende de laatste 2 jaren. Kinderen die momenteel deelnemen in de eerste FTI-behandeling zullen de kans krijgen om zich in te schrijven voor deze driedubbele behandeling wanneer zij aankomen voor hun laatste bezoek van de huidige behandeling.
Dit staat de kinderen toe om de FTI’s verder in te nemen zonder een dosis te moeten missen.
Een blik op de medicijnen : Pravastatin (op de markt gebracht als Pravachol or Selektine) : dit wordt gewoonlijk gebruikt voor het verminderen van de cholesterol en het voorkomen van cardiovasculaire ziektes. Zoledronic acid is een bisphosphonate : wordt gewoonlijk gebruikt als een “botmedicijn” voor het verbeteren van osteoporose en om skeletachtige breuken te voorkomen bij mensen die aan sommige vormen van kanker lijden. Lonafarnib is een FTI (Farnesyltransferase inhibitor) : een medicijn die een abnormaliteit in progeriacellen in het laboratorium kan omkeren en de ziekte heeft verbeterd bij progeriamuizen. De drie medicijnen blokkeren de productie van de farnesylmolecule dat pogerin nodig heeft om de ziekte Progeria tot stand te brengen.
De patiënten zullen telkens om de 6 maanden en dit gedurende een periode van 2 jaar naar Boston reizen voor testen en onderzoeken die tussen de 4-7 dagen duren. Voor de enige FTI-behandeling moesten de kinderen naar Boston reizen om de 4 maanden. In Maart 2009 namen vijf kinderen van de leeftijd tussen 2-3 jaar deel aan een haalbaarheidsstudie van één maand om te bepalen of de bijwerkingen van de drie medicijnen samen verdraaglijk/aanvaardbaar waren. De resultaten waren positief en baanden zo een weg voor de volle twee jaar durende driedubbele behandeling waarop 45 kinderen met Progeria zich kunnen inschrijven. Hoed af voor deze verbazingwekkende families !
D) June 2011: PRF-gefundeerde studie identificeert Rapamycin als mogelijke behandeling tegen progeria : @Progeria Research Foundation
Onderzoekers van het ‘National Institutes of Health’ en het ‘Massachusetts General Hospital’ in Boston, MA publiceerde vandaag een nieuwe studie in Science, Translational Medicine dat kan leiden tot een nieuwe medicamenteuze behandeling voor kinderen met Progeria . 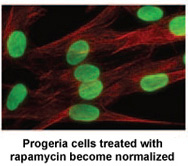 Rapamycine is een FDA goedgekeurd geneesmiddel dat reeds eerder aantoonde de levens van niet-progeria muismodellen te verlengen. Deze nieuwe studie toont aan dat Rapamycine de hoeveelheid van de ziekte-veroorzakende eiwit progerin met 50% vermindert, de abnormale nucleaire vorm verbetert en de levensduur van Progeriacellen verlengt. Deze studie levert het eerste bewijs dat Rapamycine in staat zou kunnen zijn om progerin’s schadelijke effecten in kinderen met Progeria te verminderen.
Rapamycine is een FDA goedgekeurd geneesmiddel dat reeds eerder aantoonde de levens van niet-progeria muismodellen te verlengen. Deze nieuwe studie toont aan dat Rapamycine de hoeveelheid van de ziekte-veroorzakende eiwit progerin met 50% vermindert, de abnormale nucleaire vorm verbetert en de levensduur van Progeriacellen verlengt. Deze studie levert het eerste bewijs dat Rapamycine in staat zou kunnen zijn om progerin’s schadelijke effecten in kinderen met Progeria te verminderen.
Klik hieronder voor links naar de media verhalen
De Progeria Research Foundation was blij te kunnen voorzien in cellen van de PRF Cell & Tissue Bank, voor dit project, en helpen dit onderzoek verder te financieren door middel van subsidieprogramma’s. Deze opwindende nieuwe studie toont het opmerkelijke tempo naar progeria onderzoek, en biedt verder inzicht in het verouderingsproces dat ons allemaal aangaat. *”Rapamycin Reverses Cellular Phenotypes and Enhances Mutant Protein Clearance in Hutchinson-Gilford Progeria Cells” Kan Cao, John J. Graziotto, Cecilia D. Blair, Joseph R. Mazzulli, Michael R. Erdos, Dimitri Krainc, Francis S. Collins 29 June 2011 Vol 3 Issue 89 Epub ahead of print De Standaard : klik hier
23.08.2014 – 31.08.2014 : Children’s Hospital Boston – 23.08.2014 – 31.08.2014
Onze Michiel is sinds 2007 onafgebroken bezig met de behandeling en onze Amber sedert 2009.
Michiel en Amber zijn in augustus 2014 nog naar Boston geweest en moeten nu 1 x per twee jaar naar Amerika, gedurende een periode van week voor diverse onderzoeken : röntgenfoto’s, hartonderzoek, gewicht, lengte, bloeddruk, enz. / eventuele aanpassing van de dosis en men krijgt dan een nieuwe voorraad mee voor de eerstvolgende 2 jaar.
Waar staan we nu :
De “Triple Trial” is verlengd voor 2 jaar, en uitgebreid tot 80 kinderen, zodat elk kind de toegang krijgt tot een behandeling die hen langer en gezonder zou kunnen laten leven. Echter, tijdens deze laatste testfase, nemen alle kinderen alleen het medicijn Lonafarnib terwijl het team het proces analyseert van de vele duizenden data-elementen (elk kind onderging meer dan 100 tests per bezoek!) van de triple-medicijnenkuur : dit kan echter jaren duren.
Trials history at-a-glance:
De FTI ‘Lonafarnib’ is bewezen als behandeling voor Progeria. In 2012 werden de studieresultaten gepubliceerd en blijkt dat er bij elk kind een verbetering was in één of meer gebieden, waaronder het vitale cardiovasculaire systeem. En in mei 2014, uit verdere studie bleek dat Lonafarnib (en eventueel de andere 2 drugs getest in de Triple Trial) de geschatte levensduur verhoogt met 1,6 jaar (de tijd zal leren of dit nog zal toenemen).
Trial 4
is in april 2016 begonnen en loopt nog. Het is een 2-drug, fase 1-studie om de maximaal getolereerde dosis (MTD) van het geneesmiddel Everolimus te bepalen, die samen met de behandeling Lonafarnib wordt gebruikt. Zodra de MTD is bepaald, financiert en co-coördineert PRF de fase 2-portie, die de effectiviteit van de combinatie van 2 geneesmiddelen zal testen.
Nieuw medicijn, nieuwe hoop voor kinderen met Progeria: Fase 1, 2-geneesmiddelenstudie begint in april 2016
PRF is verheugd om aan te kondigen dat we nu een nieuwe klinische proef financieren en co-coördineren, die een combinatie van twee geneesmiddelen van Lonafarnib plus Everolimus zal beoordelen. Everolimus is een vorm van het middel Rapamycine, maar Everolimus kan gemakkelijker aan de kinderen met Progeria worden gegeven omdat het minder bloedafnames vereist om het medicijnniveau te meten. Hoewel Lonafarnib de ontwikkeling van progerine kan blokkeren, lijkt Rapamycine het mogelijk te maken dat cellen sneller het toxische progerine verwijderen. Dus met Rapamycine gericht op een andere weg dan lonafarnib, kan de combinatie een “een-tweetje” blijken te zijn voor Progeria – hopelijk een betere behandeling dan lonafarnib op zichzelf.
Rapamycin is een door de FDA goedgekeurd medicijn waarvan eerder is aangetoond dat het de levensduur van niet-Progeria-muismodellen verlengt. Een onderzoek * door onderzoekers van de NIH in Bethesda, MD en het Massachusetts General Hospital in Boston toont aan dat Rapamycine de hoeveelheid van het ziekteveroorzakende eiwit progerine met 50% verlaagt, de abnormale kernvorm verbetert en de levensduur van Progeria-cellen in het laboratorium.
Rapamycine staat bekend om zijn anti-aging eigenschappen bij muizen. Deze bevindingen maken deel uit van een groeiende lijst van onderzoeken die helpen de theorie te valideren dat het vinden van de remedie voor Progeria ook de gehele verouderende bevolking ten goede kan komen.
* K. Cao, J. J. Graziotto, C. D. Blair, J. R. Mazzulli, M. R. Erdos, D. Krainc, F. S. Collins, “Rapamycin Reverses
Cellulaire fenotypes verbeteren de mutante eiwitafscheiding in Hutchinson-Gilford Progeria-syndroomcellen. “Sci. Vert. Med. 3, 89ra58 (2011). Het Boston-testteam begint met fase 1, waar het onderzoeksteam de veiligste maximale dosis Everolimus voor kinderen met Progeria zal bepalen. Het team begint met het geven van een zeer lage dosis van de medicatie aan drie kinderen en observeert zorgvuldig op bijwerkingen. Als de toxiciteit minimaal is, nemen nog eens drie kinderen deel aan een hogere dosis van het geneesmiddel. Dit patroon wordt herhaald tot de veiligste maximale dosis Everolimus is vastgesteld en de volgende onderzoeksfase kan beginnen. Tijdens fase 2 zal het onderzoek bepalen of de effecten van de combinatie van twee geneesmiddelen op de ziekte beter zijn dan alleen Lonafarnib. Samen kunnen deze Phase 1-2-behandelingsstudie tot 80 kinderen inschrijven en een geschatte 3,5-4 jaar nodig hebben om voltooid te worden, voor een bedrag van $ 2,5 miljoen dollar.
De Progeria Research Foundation leverde cellen voor dit project van de PRF Cell & Tissue Bank en hielp het onderzoek financieren via ons subsidieprogramma – meer bewijs dat PRF’s onderzoeksgerelateerde programma’s essentieel zijn voor vorderingen in de richting van de remedie. Deze nieuwe proef is een gezamenlijke inspanning die voortbouwt op de kennis die is opgedaan met de eerdere Progeria-onderzoeken. De kinderen zullen worden gezien door vrijwel hetzelfde team van artsen uit het Boston Children’s Hospital, het Dana-Farber Cancer Institute en Brigham and Women’s Hospital, die nu allemaal de wereldberoemde expertise hebben in Progeria, evenals de medicijnen die erbij betrokken zijn.
Anno augustus 2017 :
Michiel en Amber zijn in augustus 2017 nog naar Amerika geweest en zijn daar beiden gestart met de nieuwe kuur, die 2 jaar zal duren.
Wat zijn de meer recente ontwikkelingen in het Progeria-onderzoek volgens Progeria Research Foundation?
Bij het nastreven van onze missie om aanvullende behandelingen en de remedie voor Progeria te ontdekken, financiert PRF geavanceerde onderzoeksinitiatieven die geneesmiddelen en methoden onderzoeken die zich richten op de verschillende routes tijdens de progressie van de ziekte van Progeria. Die gebieden hebben betrekking op: 1) het corrigeren van de LMNA-mutatie door genoombewerking; 2) remming van de productie van progerine-mRNA met RNA-therapeutica; en 3) het beïnvloeden van progerine-eiwit, of eiwitten die een interactie aangaan met of stroomafwaarts van progerine werken met behulp van geneesmiddelen die kleine moleculen worden genoemd.
1) Gentherapie : In januari 2021 publiceerde het wetenschappelijke tijdschrift NATUUR baanbrekende resultaten die aantonen dat genetische bewerking in een muismodel van Progeria de Progeria-mutatie in veel cellen corrigeerde, verschillende belangrijke ziektesymptomen verbeterde en de levensduur van de muizen met 240% verlengde. [8] Aanvullende preklinische studies zijn nodig om deze resultaten te onderzoeken, waarvan we hopen dat ze op een dag zullen leiden tot een klinische proef.
“Het zien van deze dramatische reactie in ons Progeria-muismodel is een van de meest opwindende therapeutische ontwikkelingen waar ik in 40 jaar als arts-wetenschapper deel van heb uitgemaakt”, zegt Francis Collins, MD, PhD, directeur van de National Institutes of Health .
2) RNA Therapeutics : In maart 2021 heeft PRF bijgedragen aan twee zeer opwindende doorbraakstudies over het gebruik van RNA-therapeutica in Progeria-onderzoek, die beide proberen het vermogen van het lichaam om de productie van progerine op RNA-niveau te produceren, te blokkeren. Eén studie toonde aan dat de behandeling van Progeria-muizen met een medicijn genaamd SRP2001 de schadelijke progerine-mRNA- en eiwitexpressie in de aorta, de belangrijkste slagader in het lichaam, en in andere weefsels verminderde. Aan het einde van de studie vertoonden de muizen een verhoogde overleving van meer dan 60% [9]De andere studie toonde een reductie van 90 – 95% van het toxische progerine-producerende RNA in verschillende weefsels na behandeling met een medicijn genaamd LB143. Progerine-eiwitreductie was het meest effectief in de lever, met aanvullende verbeteringen in het hart en de aorta. [10]
3) Het progerine-eiwit richten door middel van de ontwikkeling van kleine moleculen : een medicijn genaamd progerinine is veelbelovend gebleken. In een Progeria-muismodel verhoogde progerinine het lichaamsgewicht en verlengde het de levensduur met 10 weken, een aanzienlijke doorbraak, vergeleken met een verlenging van de levensduur van twee weken bij met lonafarnib behandelde muizen. [11] Een ander molecuul met een opwindend potentieel richt zich op het pro-inflammatoire cytokine interleukine 6 (IL6) door tocilizumab opnieuw te gebruiken, een medicijn dat is goedgekeurd voor de behandeling van menselijke artritis. Progeroid-muizen behandeld met tocilizumab vertoonden huidverbeteringen, gewichtstoename, betere locomotorische activiteit en verlenging van de levensduur.
Het veld van Progeria-onderzoek boekt grote vooruitgang en groeit voortdurend in omvang en verfijning terwijl de zoektocht naar effectieve behandelingen en genezing doorgaat. Toegewijde, ijverige wetenschappers leiden het veld naar doorbraken en nieuwe behandelingen die kinderen met Progeria helpen langer en gezonder te leven, terwijl ze ook de ontdekking van hartaandoeningen en veroudering stimuleren.
12. Klassieke en Niet-klassieke Progeria
Kinderen met klassieke HGPS lijken sterk op elkaar. Er is een groep van patiënten met Progeria die naast de Progeriasymptomen ook nog een aantal symptomen vertonen die lijken op mandibulo-acrale dysostosis (MAD).De verklaring hiervoor is dat MAD net als Progeria veroorzaakt wordt door een slecht functioneren van Lamine A.De symptomen variëren in ernst en duur.Omdat zij wel een vroegtijdige veroudering tonen, en uiteindelijk alle kenmerken krijgen die bij Progeria voorkomen maar wel in een ander tempo, noemt men het niet-klassieke Progeria. niet-klassieke Progeria De verschillen met klassieke Progeria zijn:
- De groei blijft minder achter: Kinderen met niet-klassieke Progeria worden1,30 mtot1,45 m, terwijl Progeria-patiënten zelden groter worden dan1,15 m.
- Het hoofdhaar blijft veel langer aanwezig en verdwijnt niet volledig op oudere leeftijd.
- Verlies van onderhuids vet vindt minder snel plaats, en de wangen en de huid onder de kin behouden langer hun onderhuids vet. Dit verdwijnt pas op volwassen leeftijd.
- Botverlies is duidelijker dan bij Progeria, en aanwezig in vele beenderen zoals schedeldak, kaak, sleutelbeen, vingerkootjes, en ribben. Het treedt ook op aan de botten die het aangezicht vormen (viscerocranium) maar wel pas later, op volwassen leeftijd. Bij veel botverlies bestaat er een toegenomen risico op botbreuken. Deze kinderen krijgen die dan ook vaak, veelal vanaf de leeftijd van3 a4 jaar.
- Een zekere mate van bloedverwantschap in het huwelijk van de ouders blijkt meer voor te komen (in het onderzoek van Dr. Hennekam 4 van de 13 families)
- De kans op het bereiken van de volwassen leeftijd is groter dan bij Progeria.
Congenitale (aangeboren) Progeria
- Er is een klein aantal kinderen dat al bij de geboorte symptomen van Progeria laat zien. Zij hebben een laag geboortegewicht, afwezigheid van onderhuids vetweefsel, weinig haar, botverlies van teen- en vingerkootjes, niet sluiten van de schedelnaad (zichtbaar in wijde naden en vergrote fontanellen), smal gezichtje met licht uitpuilende ogen en kleine kin, botverlies in uiteinden sleutelbeenderen. Minder opvallende kenmerken zijn: afwezige oorlelletjes, kleine brede nageltjes, prominente gewrichten, opvallend grote aderen op het hoofd en een verharde dunne huid (scleroderma). Al deze kenmerken kunnen bij aangeboren Progeria al bij de geboorte zichtbaar zijn. Het is waarschijnlijk dat bij deze kinderen er een bijzondere oorzaak is voor het niet goed functioneren van Lamine A, maar dat is tot op heden nog niet onderzocht.
Verwante ziektes De oorzaak van Progeria ligt op chromosoom 1, bij het gen dat betrokken is bij de opbouw van de Laminelaag. Dat geldt ook voor een aantal verwante ziektes:
partiële lipodystrofie, een ziekte waardoor er een tekort aan onderhuids vetweefsel ontstaat.
musculaire dystrofie, Emery-Dreifuss musculaire dystrofie (EDMD), komt voornamelijk bij jongens voor, en Limb-girdle musculaire dystrofie (LGMD), een vorm van Emery-Dreifuss musculaire dystrofie.
Charcot-Marie-tooth , oftewel CMT, is een van de meest voorkomende neurologische ziekten, meestal niet levensbedreigend.
Mandibulaire Acrale Dysplasie (MAD), is een zeldzame autosomale recessieve ziekte. Patiënten met MAD vertonen veel overeenkomsten met progeriapatiënten. Wel worden MAD-patiënten gemiddeld een stuk ouder dan progeriapatiënten.
Werner syndroom: Het Wernersyndroom is ook een ouderdomssyndroom. Bij dit syndroom, dat erfelijk is, treden er ouderdomsverschijnselen rond het begin van de puberteit. De gemiddelde levensverwachting is 20-30 jaar.
RD, restrictieve dermopathie: Neonatale Progeria, een zeer ernstige, zeldzame ziekte. Deze kindjes overlijden vaak al in de eerste weken na de geboorte.
13. Organisaties : Website: Progeria Family Circle http://www.progeria.nu/index2.html In 1996 realiseerden de ‘progeriafamilies’ uit Europa zich hoe belangrijk het is voor de patiëntjes en hun gezinnen om elkaar te kunnen ontmoeten. Tijdens een reünie kan men andere families en artsen om raad en advies vragen, specifieke problemen bespreken. Gesterkt door de ervaring dat onderling contact een bron is voor meer levenskwaliteit, stichtten betrokken families in Europa in 1997 de Progeria Family Circle, een Europese stichting voor kinderen met Progeria.De Progeria Family Circle (stichting voor kinderen met Progeria) wordt gevormd door alle ouders en betrokkenen van kinderen met deze ziekte. Het is in feite de naam voor een ideaal: zij willen elkaar helpen.
Als ‘ervaringsdeskundigen’ proberen ze klaar te staan voor elkaar. Door de aard van de ziekte willen zij daarnaast ook nog wat speciaals doen: bijeenkomsten organiseren. Daarvoor is geld nodig, en dan kan je in onze wereld niet zonder officiële statuten, banken en besturen. Vandaar dat er een Europese stichting voor kinderen met Progeria (het syndroom van Hutchinson-Gilford) is opgericht: de Progeria Family Circle. Maar het geld op de rekening van de Progeria Family Circle is van en voor alle ouders. Omdat de gezondheidszorg in veel Europese landen noodzakelijke aanpassingen (bv. aan het huis, een speciale stoel ed.) al vergoed, is dat geld bestemd voor het organiseren van bijeenkomsten.
Ze betalen niemand een salaris, iedereen werkt als vrijwilliger mee aan het ideaal van een beter leven voor deze kinderen en hun ouders. Voor persoonlijk contact met de Progeria Family Circle kunt u zich wenden tot : Marjet Stamsnijder PROGERIA FAMILY CIRCLE Stichting voor kinderen met Progeria Nude 6 3911 VK Rhenen Nederland Telefoonnummer: +31 (0)317 615391 E-mail adres: progeriafamilycircle@hetnet.nl
Nieuwe Progeria-families weten deze stichting te vinden en zij hopen alle ‘ nieuwe’ families te vinden. De warme respons die zij tot nu toe mochten ervaren sterkt hun motivatie voor de toekomst.
The Sunshine Foundation : www.sunshinefoundation.org Bill Sample runt een wellfare organisatie, nl. de Sunshine Foundation, die zich (net als alle andere soortgelijke stichtingen) aan regels moet houden. Deze organisatie doet aan “Last wish forfilling” en helpt kinderen met AIDS, Progeria en andere chronische ziekten om hun wensen en dromen waar te maken. Het is te vergelijken met de “Make-a-Wish Foundation”. Ze hebben tot nu toe ongeveer 22.000 kinderen geholpen. In 1981 heeft de “Sunshine Foundation” 2 Progeriakinderen samen gebracht, namelijk Meg uit de Verenigde Staten en Francy uit Zuid-Afrika. Sindsdien worden er jaarlijks zulke ontmoetingen georganiseerd.
Deze jaarlijkse reünie, die een week duurt, is het hoogtepunt in het leven van de tot nu toe 45 gekende Progeria kinderen. Zij gaat steeds door in de Verenigde Staten. Hier kunnen ouders en kinderen elkaar ontmoeten, ervaringen uitwisselen, specifieke problemen bespreken en vriendschappen sluiten. De betrokken families vinden bij elkaar de informatie en hulp die zij zo hard nodig hebben bij het vervullen van hun moeilijke levenstaak. Dit jaarlijkse onderlinge contact is een bron voor meer levenskwaliteit van deze kinderen en dit is enkel mogelijk dankzij de Sunshine Foundation.
Progeria Research Foundation : www.progeriaresearch.org Deze organisatie in de VS stelt zich ten doel progeriapatiëntjes te helpen. Deze stichting richt zich vooral op het werven van fondsen om wetenschappelijk onderzoek mogelijk te maken teneinde de oorzaken van de vroege veroudering bij Progeria te vinden.
Hutchinson-Gilford Progeria Syndrome Resource Center : www.hgps.net Eén van de eerste sites met informatie over Progeria. Dit is een bron van foto’s, connecties en een discussie forum Europrogeria : www.europrogeria.org Bij Europese artsen is er veel welwillendheid en inzet om in onderlinge samenwerking verder te zoeken naar meer inzicht in de basis van het syndroom. In 2003 richtten zij een consortium op: “EUROPROGERIA”. Er worden medische congressen georganiseerd waar wetenschappers uit de hele wereld hun expertise onderling uitwisselen en er relaties worden gelegd met aan Progeria verwante syndromen.
14. Besluit :
Wij willen graag besluiten met volgende woorden :Michiel is humoristisch, spontaan, zelfzeker, eigenzinnig, enthousiast, sociaal, koppig maar een geweldig kind. Hij heeft zich ontwikkeld tot een zelfbewust en vrolijk kind, ondanks zijn zeldzame ziekte. Hij gaat naar school en ondanks zijn eigenzinnige persoonlijkheid heeft hij altijd vriendjes en vriendinnetjes om zich heen, die hem volledig accepteren. Hoe kan het ook anders. Het is een kind boordevol energie en humor en met zijn levensvreugde maakt hij veel indruk op de mensen, ook op vreemden. Hij is de trots van ons allemaal. Amber is een schattig meisje die ons tot nu toe al veel vreugde en plezier heeft bezorgd. Ze is heel speels, heel guitig en heel hartveroverend. Michiel en Amber zijn de zon en het vuur (warmte) in ons leven. Zij hebben ons de werkelijke betekenis van het leven leren kennen : vreugde, liefde, samenhorigheid en vooral verdraagzaamheid voor het “anders-zijn”. ! MICHIEL EN AMBER DRAGEN ENORM BIJ AAN ONZE LEVENSKWALITEIT !
Click here First-Ever progeria clinical drug trial reaches 1 year: Click here Announcing the Progeria Triple Drug Trial: Click here